Quirónsalud







Blog del Dr. Daniel Martín Fernández-Mayoralas. Neurología. Complejo Hospitalario Ruber Juan Bravo y Hospital Universitario Quirónsalud Madrid

- 202410abr
Síndrome cognitivo afectivo del cerebelo: Un diagnóstico a tener en cuenta.
El cerebelo es una región del encéfalo, situado a modo de "mochila" en la fosa craneal posterior, detrás del tronco del encéfalo.
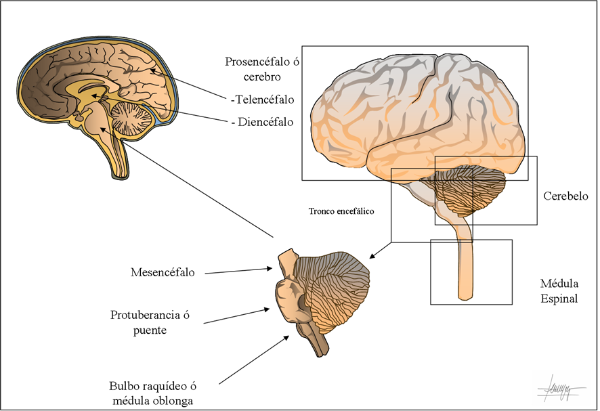
Su función principal es de integrar las vías sensitivas y las vías motoras. La participación del cerebelo en procesos motores a través de esta integración, mediante la coordinación del movimiento, es conocida desde hace no menos de doscientos años. Sin embargo, no es hasta bien entrado el siglo XX, cuando algunos científicos involucran al cerebelo funciones diferentes, no motoras, como el procesamiento cognitivo o las emociones.

Existe un patrón de anomalías denominado síndrome cerebeloso cognitivo afectivo, que incluye deficiencias de las funciones ejecutivas (planificación, fluidez verbal, memoria de trabajo, déficit de atención: esto es, muchas de las funciones afectadas en niños con trastorno de atención con/sin hiperactividad -TDAH-), disfunción visoespacial, cambios de personalidad, embotamiento del afecto, tristeza, desinhibición o comportamiento inapropiado y diversas dificultades del habla. Es importante conocer que la baja velocidad de procesamiento, clínica y neuropsicológica es prácticamente constante, aunque los síntomas y signos presentes dependerán del área o áreas del cerebelo y sus conexiones con otras regiones encefálicas involucradas.
Este síndrome fue descrito por primera vez por Schmahmann y Sherman en 1998. Puede ser secundario a numerosos trastornos diferentes que afecten al cerebelo, como un tumor benigno cerebeloso y/o como secuela de su extirpación quirúrgica, una afectación vascular (más frecuente el recién nacido prematuro), entre otras, siendo la secuela (transitoria o crónica) de la ataxia cerebelosa aguda con o sin cerebelitis (la causa más común de ataxia aguda en la infancia y la adolescencia), habitualmente precedida de una infección viral, la causa más frecuente del síndrome cognitivo afectivo del cerebelo.
A pesar de su descripción clara y su confirmación como entidad, es relativamente desconocida para profesionales, pacientes y familiares. Es importante evitar el tópico de que las afectaciones cerebelosas se limitan a disfunciones motoras y de coordinación y detectar la presencia de los problemas psiquiátricos y cognitivos propios del síndrome (completo o incompleto), de cara a un tratamiento adecuado pedagógico, psicológico y farmacológico (que puede ser eficaz en numerosos casos).
- 202421feb
Síndrome de Pitt-Hopkins
Por su interés, traducimos al español este ensayo publicado en la revista BRAIN, obra de ciencia ficción basada en las experiencias de los padres de niños con el síndrome de Pitt-Hopkins, una enfermedad genética extremadamente rara causada por una mutación de novo o eliminación en el gen del factor de transcripción TCF4, que organiza el tejido neural durante el desarrollo embrionario. El autor es Brian Routledge, padre de un niño llamado Chris, afecto del síndrome de Pitt-Hopkins.

Nacido en las profundidades del Sol, el neutrino brotó, expulsado por una enorme llamarada solar, y se dirigió hacia el espacio, donde ocho minutos después cambiaría el mundo de alguien. Atravesó el núcleo de Mercurio indemne, evitó a Venus que se encontraba, en ese momento, en el otro lado del Sol, y se sumergió en la frágil atmósfera de un planeta azul y blanco. Sin obstáculos por la materia sólida, pasó sin esfuerzo a través del aire, las tejas del techo, las paredes de ladrillo y el suelo de madera de la casa hasta que encontró materia orgánica. Se deslizó a través de los intersticios moleculares del tejido blando y chocó contra la doble hélice de una célula a punto de dividirse, antes de continuar su camino eterno.
Las enzimas de reparación de ácidos nucleicos neonatales inmaduros no detectaron el daño al gen de control vital que, entre otras funciones, organizaba el desarrollo neural embrionario.
Los nuevos padres colocaron al niño dormido en la cuna recién construida. Su abrazo apretado y el tierno beso hablaban de su alegría, esperanzas y expectativas. Las sospechas de la madre surgieron antes, pero la revisión del desarrollo al año confirmó sus peores temores. Siguieron la incredulidad, la ira, la confusión y el dolor, de los cuales surgió una determinación firme de brindar la mejor vida posible a su hijo y maximizar su potencial. Se exploraron todas las posibilidades, y años de terapia intensiva se tradujeron en mejoras en el desarrollo.
Se logró caminar, pero tarde. Se estableció un nivel de comunicación, aunque no verbal. Actos simples como sostener una cuchara y alimentarse a sí mismos se celebraban como si fuera Navidad. Los padres y hermanos gradualmente reconocieron y aprendieron a leer y comprender la personalidad y el carácter de su hijo, mientras emergía del caos genético que el neutrino había creado.
Pero se alcanzó un punto en el que parecía que se lograba poco o ningún progreso adicional. La familia amaba al niño por quien era, no por cómo era.
El hombre con el traje oscuro colocó el expediente en la mesa de la conferencia, junto a sus gafas, y se recostó, estirando los músculos del cuello y los hombros. Las caras expectantes de quienes estaban reunidos se centraron en él, esperando su reacción y respuesta. "Así que funciona: reparación genómica completa y resecuenciación", dijo. Las cabezas asintieron. "¿No hay efectos secundarios o consecuencias ocultas?" "No que hayamos identificado", dijo el hombre con bata blanca al otro extremo de la mesa. "Los animales mostraron una restauración completa de la función del producto génico. Las pruebas metabólicas y de comportamiento no parecen desviarse de las normas de control establecidas".
El hombre hizo una pausa, sumido en sus pensamientos antes de anunciar: "Se han acordado las bases para la creación de la empresa derivada y se están elaborando los documentos necesarios. Recibirán los contratos cuando estén listos. Esto pondrá cierta distancia entre la universidad como institución de investigación y el desarrollo de los aspectos comerciales". Miró a los demás por encima del borde de las gafas que descansaban en su nariz.
"Damas y caballeros, les felicito. Estamos al borde de una nueva era en la que las enfermedades genéticas raras serán eliminadas para siempre del genoma humano. Sus nombres estarán junto a los de Mendel, Crick, Watson, Franklyn, Charpentier y Doudna en el panteón de los Héroes Genéticos". Se puso de pie.
La tos del hombre con la bata blanca atrajo la atención del Hombre de Negro.
"Si me permiten decir algo", comenzó. "Estoy de acuerdo con sus afirmaciones sobre el futuro de las terapias génicas, pero los resultados del tejido de la línea germinal no son tan convincentes como para las células somáticas. Hemos documentado células de línea germinal no funcionales y, en algunos casos, incluso aberrantes. La técnica, en un pequeño número de casos, parece introducir errores adicionales en la línea germinal. Recomendaría, en este momento, si me permiten ser tan audaz de aconsejar, que lo mejor que podemos ofrecer es una corrección de una sola generación".
—Entendido —dijo el hombre con el traje negro—. ¿Puede resolverse el problema?
—Con tiempo y suficiente dinero —respondió el hombre de la bata blanca. Las cabezas asintieron nuevamente.
—Bien, da el siguiente paso —dijo el Hombre de Negro sobre su hombro mientras salía de la habitación.
Las caras reunidas se dirigieron ahora hacia el otro extremo de la mesa de conferencias. "Lo que necesitamos ahora", observó el Científico Jefe, "es un candidato adecuado. ¿Alguna idea?"
El anuncio hizo que las ondas del aire y la ciberesfera zumbasen de actividad. Se convirtió en el primer tema de las versiones matutina, de mediodía y nocturna de las noticias televisadas. Reporteros de las estaciones de noticias de 24 horas buscaron a cualquier científico, líder religioso o filósofo dispuesto a dar un comentario y/o una opinión. Las líneas telefónicas para programas de radio con participación del público se iluminaron intensamente cuando el tema se convirtió en el último centro de debate. Organizaciones benéficas y grupos de apoyo en línea para enfermedades raras, y algunas no tan raras, se vieron inundados de solicitudes de más información sobre dónde y cuándo estaría disponible la técnica.
Ningún padre renuncia nunca a la esperanza para su hijo y la promesa de una cura inmediata abrió una antigua herida, incluso en familias que habían aprendido a aceptar a su hijo, discapacidades incluidas. El Dorado de la normalidad, la Tierra Prometida, la Utopía donde su hijo ahora podría convertirse en el individuo que habían deseado y anhelado en un principio, parecía estar a punto de hacerse realidad.
Hubo un proceso de selección inicial que incluía la secuenciación del gen dañado del niño, seguido de un análisis psicológico exhaustivo de los padres, sondeando su motivación para considerar el procedimiento para su hijo y evaluando sus capacidades para hacer frente, a corto y largo plazo, a las circunstancias familiares cambiadas que surgirían. Se brindó capacitación mediática para preparar a la familia para el escrutinio público que inevitablemente resultaría cuando se hiciera pública la noticia. Se crearon páginas de financiación colectiva, se hicieron apelaciones y se organizaron eventos patrocinados para proporcionar la financiación necesaria, y donantes adinerados famosos contribuyeron a cambio de un reconocimiento público.
El hombre con el traje negro dejó el informe. Los presentes esperaban con la respiración contenida.
—En opinión de los expertos, el niño no tiene la capacidad de entender ni la capacidad de comunicarse más allá de unos pocos signos simples. No hay indicación detectable de funciones cognitivas superiores. Concluyen que el niño no puede otorgar un consentimiento informado. La decisión final queda en manos de los padres.
Se tomó tiempo para preparar todo, pero finalmente llegó el día para administrar la terapia. El Científico Jefe y el hombre con el traje negro se habían escapado de la abarrotada conferencia de prensa y ahora estaban de pie fuera de la sala de tratamiento. Dentro, los padres se tomaban de las manos y hablaban en voz baja al niño como distracción del dolor causado por la inserción del tubo en una vena a través del cual se administraría el agente de reparación y resecuenciación. La enfermera conectó el tubo a la bolsa, abrió la válvula y apretó suavemente la bolsa. La audiencia reunida permaneció en silencio mientras las primeras gotas se formaban en la línea intravenosa, se transformaban lentamente en un flujo y finalmente fluían hacia el brazo del niño sujeto a la cama. Se desató un aplauso.
Tengo miedo. Algo está sucediendo. Sentí la preocupación de mis padres esta mañana y luego me trajeron aquí y ahora no puedo moverme. Realmente no me gustan los hospitales, los olores y la iluminación me hacen sentir un poco asustado. Recuerdo que si estoy en un hospital, me suceden cosas malas, como ahora que me clavaron una aguja en el brazo. No me siento mal y no tengo dolor. Me han hecho muchas preguntas recientemente por personas extrañas y he escuchado la palabra 'dañado' muchas veces. Realmente no sé lo que significa, pero creo que indica que soy diferente. No me siento dañado. Traté de responder sus preguntas, pero no sabían cómo hablar conmigo. Mis padres no estaban presentes, así que no pudieron preguntarme ni me han explicado por qué estoy aquí. Intento responder a todos porque me gustan las personas, pero todo es tan difícil y agotador, ya que mi cuerpo no hará lo que quiero. Puede pasar mucho tiempo antes de que mis pensamientos se conviertan en acciones. Puede llevar tiempo recordar las cosas. Sé que nunca he podido hacer muchas de las cosas que hace mi hermano porque siempre he tenido dificultades para hacer que mi cuerpo funcione correctamente. Dicen que pronto podré hacerlo porque me están 'reparando'. No siempre entiendo rápidamente todo lo que me dicen, pero he mejorado en ello y sigo aprendiendo a medida que crezco, aunque lentamente. Todavía soy un niño.
Creo que algunas personas tienen miedo de mí. Mis padres fueron impacientes al principio, pero ahora se dan cuenta y me comprenden más, así que esperan. He aprendido a pedir comida con signos simples que puedo manejar, y mi madre puede interpretar fácilmente mis estados de ánimo. No creo que esté dañado. Me enojo, me rio, sonrío, disfruto de estar vivo. Soy feliz. Amo a mis padres y sé que me aman, aunque sea diferente. No soy un monstruo ni un fenómeno. Me siento un poco extraño ahora y mi cabeza empieza a doler. Si me están reparando, ¿qué cambiará? ¿Seguiré siendo yo? ¿Seguiré disfrutando de los plátanos? ¿Incluso podré seguir hablando? ¿Mi color favorito seguirá siendo el azul? ¿Seguiré prefiriendo Star Trek a Star Wars? ¿Quizás ya no amaré a mis padres y hermanos? ¿Quizás no amarán la nueva versión de mí?
Bibliografía: https://academic.oup.com/brain/article/147/1/2/7510886
 .
.
Copyright © 2024, © The Author(s) 2023. Publicado por Oxford University Press en nombre de Guarantors of Brain.
- 20241feb
Tratamiento Cognitivo Conductual en adolescentes con Trastorno por Déficit de Atención con/sin Hiperactividad (TDAH)
Los padres nos preguntan con frecuencia sobre la importancia del tratamiento cognitivo conductual en pacientes con TDAH. Hemos de decir que en la mayoría de los casos es muy importante.

El TDAH persiste con frecuencia en la adolescencia y, aunque los medicamentos (por ejemplo, los estimulantes como el metilfenidato o la lisdexanfetamina o los no estimulantes como la guanfacina de liberación retardada) son útiles para controlar sus síntomas, son menos efectivos para mejorar el funcionamiento en las actividades del día a día, refiriéndonos con esto, especialmente a la disfunción ejecutiva (organizar, planificar, tomar buenas decisiones, saber cómo estudiar, etcétera).
Las intervenciones existentes de tratamiento cognitivo-conductual (TCC) pueden ser utilizadas en adolescentes y adolescentes con TDAH. El TTC se basa más en principios "conductuales" que "cognitivos". Cualquier intervención de TCC centrada más en la terapia cognitiva (por ejemplo, modificar pensamientos irracionales, aumentar la autoobservación) sin incorporar simultáneamente principios de manejo de contingencias, especialmente de refuerzo, es poco probable que sea efectiva.
La inclusión de los padres en la sala de terapia puede ser beneficiosa para los adolescentes con TDAH y un trastorno internalizante comórbido (los más típicos: la ansiedad y la depresión). También en ciertos casos, es importante incluir a los padres en la terapia en casos de adolescentes con TDAH y trastorno oposicionista desafiante comórbido, así como en otros casos, deben de permanecer al margen. Cada caso debe individualizarse de forma sensata, no existe una norma válida para todos los pacientes.
Normalmente, las intervenciones de TCC deben utilizarse en conjunto con la toma de medicamentos para el TDAH. Sin ellos, el TCC es probable que no sea exitoso en la mayoría de los casos.
Bibliografía recomendada: Kevin M. Antshel, et al. https://pubmed.ncbi.nlm.nih.gov/25220089/
- 202416ene
Ejercicio y TDAH
En muchas ocasiones se nos pregunta en consulta por los posibles beneficios que tiene el ejercicio sobre el TDAH. La respuesta es que sí, que muchos. Los podríamos resumir en los siguientes puntos:

• La actividad física estructurada tiene el potencial de ser un tratamiento efectivo para el TDAH en los niños.
• Datos incontrovertibles de estudios en animales indican que el ejercicio mejora el desarrollo cerebral y la conducta de los mismos.
• Muchos estudios sugieren que la actividad física puede llevar a mejorías en el funcionamiento neuropsicológico, incluida la velocidad de procesamiento y algunas funciones ejecutivas importantes en la fisiopatología del TDAH.
• Después de realizar ejercicio moderado y vigoroso a largo plazo (es decir >5 semanas), la investigación sugiere que los comportamientos relacionados con el TDAH en niños pueden ser menos graves y se pueden observar mejorías en el rendimiento en medidas de la función neuropsicológica.
• La actividad física puede ofrecer beneficios más allá del uso de psicoestimulantes. Aunque éstos fármacos son una base importante en el tratamiento del TDAH moderado o grave, su efecto positivo puede verse incrementado, de forma sinérgica, por el ejercicio.
• Aunque claramente existen beneficios en la realización de ejercicio, durante el tratamiento el TDAH, se necesitan ensayos controlados aleatorios metodológicamente sólidos y a doble ciego para determinar cuál es la eficacia exacta de la actividad física como tratamiento para el TDAH en los niños.
Para ampliar información recomendamos la siguiente revisión: https://www.sciencedirect.com/science/article/abs/pii/S1056499314000376?via%3Dihub
- 20248ene
Empoderando a los Docentes: Estrategias para detectar y abordar dificultades en el aula. (III)
Finalizamos nuestra charla con la responsable de Departamento de orientación del Colegio Europeo de Madrid: Esmeralda Velasco Espinosa (EV). Nuestro objetivo es debatir sobre cómo podemos mejorar la detección y abordaje, en el medio escolar, de los diversos trastornos específicos del aprendizaje, como los de la lectura, los trastornos del espectro autista (TEA) y el TDAH (trastorno por déficit de atención con/sin hiperactividad), como paradigmas de los trastornos del neurodesarrollo.

DM: ¿crees que es importante la evaluación neuropsicológica, así como de otros profesionales de la psicología, logopedia o pedagogía, además de por los médicos especialistas en neurología o psiquiatría infanto-juvenil?
EV: La observación cuidadosa de estas características debe ir de la mano con la consulta a profesionales especializados, como psicólogos/orientadores educativos o especialistas en necesidades especiales. La detección temprana y la colaboración con expertos permiten una intervención más efectiva y adaptaciones apropiadas con el fin de garantizar su inclusión u éxito académico. Estas medidas son importantes por varias razones como pueden ser la equidad en la educación, ya que las adaptaciones ayudan a nivelar el nivel en el que se encuentran, brindando a todos los estudiantes, independientemente de sus dificultades, la oportunidad de participar y aprender de manera significativa.
Por otro lado, facilitan el fomento de la autoestima sana, ya que realizar adaptaciones permite a los estudiantes que se enfrenten a los desafíos académicos de manera más efectiva, lo que contribuye a fortalecer su autoestima y confianza en sí mismos.
Con las dos medidas propuestas anteriormente, se mejora el rendimiento académico ya que, al ajustar el entorno de aprendizaje y las actividades para adaptarse a las necesidades individuales de los estudiantes, se facilita su participación y comprensión, lo que puede mejorar su rendimiento escolar.
La aplicación de medidas educativas inclusivas facilita el desarrollo de habilidades sociales, por ejemplo, en el caso del alumnado TEA, ya que entre las adaptaciones previstas también se pueden incluir estrategias para fomentar las habilidades sociales y la interacción exitosa con sus compañeros.
Sólo a través de la detección y la aplicación de las medidas adaptativas oportunas, se conseguirá la prevención de estigmas asociados a los distintos trastornos, aprendiendo a valorar la diversidad neurocognitiva, y a su vez, estaremos preparando a ese porcentaje del alumnado que, al igual que el resto, tendrán que enfrentarse a la etapa adulta, en la que tendrán que aplicar las habilidades necesarias para enfrentar desafíos en la vida cotidiana y, posteriormente, en el ámbito laboral.
DM: ¿Me harías un resumen final?
EV: La aplicación de medidas de adaptación es fundamental para garantizar que todos los estudiantes tengan acceso a una educación de calidad y para promover un entorno educativo inclusivo y enriquecedor. Proporcionar las medidas adecuadas a cada individuo va de la mano de la capacitación del docente para detectar, en edades tempranas, posibles dificultades que puedan limitar el acceso a la educación.
Sobre este blog
Blog sobre los temas relacionados con la neuropedciatría: déficit de atención, hiperactividad, epilepsia, cefaleas, tics, encefalitis, problemas escolares, etc.

Archivo del blog
 2.024
2.024
- 2.023
- 2.022
- 2.021
- 2.020
- 2.019
- 2.018
- Diciembre
- Noviembre
- Octubre
- Julio
- Mayo
- Abril
- Febrero
- Recomendaciones generales y específicas para el trastorno por déficit de atención/hiperactividad -TDAH- (III). Recomendaciones específicas.
- El Dr. Daniel Martín Fernández-Mayoralas ganador del “Concurso de Casos Clínicos sobre el abordaje farmacológico de pacientes con TDAH" organizado por el Grupo Saned
- Enero
- 2.017
- 2.016
Últimas entradas
- Síndrome cognitivo afectivo del cerebelo: Un diagnóstico a tener en cuenta.
- Síndrome de Pitt-Hopkins
- Tratamiento Cognitivo Conductual en adolescentes con Trastorno por Déficit de Atención con/sin Hiperactividad (TDAH)
- Ejercicio y TDAH
- Empoderando a los Docentes: Estrategias para detectar y abordar dificultades en el aula. (III)
Colaboraciones




La finalidad de este blog es proporcionar información de salud que, en ningún caso sustituye la consulta con su médico. Este blog está sujeto a moderación, de manera que se excluyen de él los comentarios ofensivos, publicitarios, o que no se consideren oportunos en relación con el tema que trata cada uno de los artículos.
Quirónsalud no se hace responsable de los contenidos, opiniones e imágenes que aparezcan en los "blogs". En cualquier caso, si Quirónsalud es informado de que existe cualquier contenido inapropiado o ilícito, procederá a su eliminación de forma inmediata.
Los textos, artículos y contenidos de este BLOG están sujetos y protegidos por derechos de propiedad intelectual e industrial, disponiendo Quirónsalud de los permisos necesarios para la utilización de las imágenes, fotografías, textos, diseños, animaciones y demás contenido o elementos del blog. El acceso y utilización de este Blog no confiere al Visitante ningún tipo de licencia o derecho de uso o explotación alguno, por lo que el uso, reproducción, distribución, comunicación pública, transformación o cualquier otra actividad similar o análoga, queda totalmente prohibida salvo que medie expresa autorización por escrito de Quirónsalud.
Quirónsalud se reserva la facultad de retirar o suspender temporal o definitivamente, en cualquier momento y sin necesidad de aviso previo, el acceso al Blog y/o a los contenidos del mismo a aquellos Visitantes, internautas o usuarios de internet que incumplan lo establecido en el presente Aviso, todo ello sin perjuicio del ejercicio de las acciones contra los mismos que procedan conforme a la Ley y al Derecho.




© 2024 Quirónsalud - Todos los derechos reservados
