Quirónsalud







Blog del Dr. Daniel Martín Fernández-Mayoralas. Neurología. Complejo Hospitalario Ruber Juan Bravo y Hospital Universitario Quirónsalud Madrid
- 201610jun
¿Cuándo debo ir al neuropediatra?
El neuropediatra es un especialista en diagnosticar y tratar las patologías específicas del sistema nervioso desde los cero a los 18 años. Es un profesional que presta especial atención al neurodesarrollo y a las interferencias que sobre éste causan diversos trastornos. Los neuropediatras atendemos un espectro muy amplio de trastornos, la mayoría de ellos leves, pero también algunos graves y complejos.

Las enfermedades o trastornos neurológicos, incluso siendo leves, causan temor y sensación de desconocimiento en los padres. Lo adecuado es consultar con el neuropediatra ante preocupaciones tales como:
- Sospecha de trastorno por déficit de atención con / sin hiperactividad.
- Retrasos en el desarrollo, global y motor.
- Trastornos de lenguaje.
- Crisis epilépticas (ausencias, convulsiones, etcétera) con o sin fiebre.
- Sospecha de retraso o discapacidad intelectual.
- Trastornos del aprendizaje escolar como la dislexia.
- Dolor de cabeza.
- Si se sospecha autismo de bajo o alto rendimiento o el llamado "síndrome de Asperger".
- Si se sospecha malformación cerebral.
- Cualquier otro síntoma, además de los mencionados, cuando pensamos que el sistema nervioso puede estar involucrado.
De cara a la consulta, especialmente la primera, es necesario darse cuenta que la mayor parte de las veces los problemas no son graves, por lo que cabe ser optimista. Aunque a veces preocupa mucho la incertidumbre, es fundamental hacer las pruebas necesarias antes de dar un diagnóstico claro si no es posible. Paciencia y entendimiento son buenos aliados.
No es mala idea preparar con antelación la visita a la neuropediatra y llevar los datos básicos: qué es lo que le pasa al paciente, desde cuándo y a qué lo atribuye y cómo ha evolucionado el problema. Es importante guardar los documentos relevantes que puedan tener relación con la visita: por ejemplo, si ya tiene test neuropsicológicos o de inteligencia en caso de sospecha de déficit de atención es conveniente llevarlos por si acaso son útiles, como también apuntar información sobre antecedentes familiares cercanos de algo similar o que tenga relación con la neurología o la psiquiatría. Según el caso es frecuente preguntar por los antecedentes del embarazo y el parto, peso del niño al nacer, talla y lo que le medía la cabeza (este dato suele venir apuntado en la cartilla). También es oportuno apuntar un boceto sobre el neurodesarollo básico: cuando empezó a sentarse, andar, a hablar, si tuvo que ir al logopeda, si hay alguna prueba antigua relacionada con la visita, etc.
El mejor tratamiento es siempre un buen diagnóstico.
420 comentarios - 202328abr
Redes sociales y salud mental
Con la colaboración de María del Rosario Campos Díaz. Psicóloga Clínica Infanto- Juvenil y responsable del Centro Cogniciona (Pozuelo de Alarcón)
Las redes sociales son aplicaciones web cuya finalidad es la comunicación entre individuos, la transmisión de información, y sabemos que la información es poder. Sin embargo, a pesar de las ventajas de su uso, por ejemplo, a nivel laboral, y teniendo en cuenta que conectarse a ellas forma parte de la conducta normal y cotidiana de la vida moderna (Andreassen, 2015), diversos estudios empíricos llegan a la conclusión de que las personas que emplean menos tiempo en redes sociales tienen mejor salud mental (Moreno y otros, 2011).

Dichas investigaciones evidencian una correlación directa entre el uso de redes sociales y la ansiedad, al igual que con la depresión. Además, está demostrado que a mayor cantidad de tiempo de uso de redes sociales, mayor probabilidad de padecer depresión y ansiedad.
El impacto en nuestro estado emocional y, más aún, en el de nuestros niños/as y adolescentes va aún más allá. Así, en el trabajo de investigación "Tiempos Modernos: Redes Sociales y Ansiedad", de Laura Vanessa García Gualdrón (asesorada por Mario Andrés Ernesto Martín Padilla), en 2015, se encontró que el uso de las redes sociales en población juvenil se encuentra caracterizada por una fuerte dependencia psicológica, e impulsividad, lo que repercute en el área social y personal de los usuarios, causando también un mal uso del tiempo.
Mucho se ha escrito ya sobre la adicción a las redes sociales, que se relaciona de forma negativa con el nivel de autoestima, y de forma significativa con el nivel de ansiedad, de manera que los estudiantes con mayor adicción a dichas redes manifiestan también un mayor nivel de ansiedad, como se constata en el estudio publicado en el siguiente artículo: Portillo-Reyes, V. Ávila-Amaya, J. A., Capps, J. W. (2021). Relación del Uso de Redes Sociales con la Autoestima y la Ansiedad en Estudiantes Universitarios. Enseñanza e Investigación en Psicología, 3(1), 139-149.
Otras consecuencias de su mal uso serían: amistades débiles y superficiales, exclusión, estrés, depresión, adicción y alteraciones en los patrones de sueño, envidia, vinculaciones irreales, entre otras (Drahošová y Balco, 2017). El uso excesivo de redes sociales también puede generar alteraciones emocionales por la visualización de información de alto impacto o de estándares y prototipos de belleza casi imposibles.
En relación a ello, señalar el artículo publicado en la revista Behavioral Psychology / Psicología Conductual, Vol. 30, Nº 3, 2022, pp. 677-691 https://doi.org/10.51668/bp.8322305s
 : EFECTO DE LA EXPOSICIÓN A IDEALES DE DELGADEZ EN LAS REDES SOCIALES SOBRE LA AUTOESTIMA Y LA ANSIEDAD, de Blanca Rodríguez-Suárez1 , José Manuel Caperos1 y José Ángel Martínez-Huertas1,2 1 UNINPSI, Universidad Pontificia Comillas; 2 Universidad Nacional de Educación a Distancia (España), que resume que el uso de redes sociales está relacionado con la aparición de trastornos de la conducta alimentaria (TCA). Los autores encuentran una diminución de la autoestima en el grupo expuesto a imágenes de carga comparativa alta y un aumento de la ansiedad. Concluyen que efecto de las imágenes sobre la autoestima está completamente mediado por el incremento en la ansiedad.
: EFECTO DE LA EXPOSICIÓN A IDEALES DE DELGADEZ EN LAS REDES SOCIALES SOBRE LA AUTOESTIMA Y LA ANSIEDAD, de Blanca Rodríguez-Suárez1 , José Manuel Caperos1 y José Ángel Martínez-Huertas1,2 1 UNINPSI, Universidad Pontificia Comillas; 2 Universidad Nacional de Educación a Distancia (España), que resume que el uso de redes sociales está relacionado con la aparición de trastornos de la conducta alimentaria (TCA). Los autores encuentran una diminución de la autoestima en el grupo expuesto a imágenes de carga comparativa alta y un aumento de la ansiedad. Concluyen que efecto de las imágenes sobre la autoestima está completamente mediado por el incremento en la ansiedad.En la actualidad, las redes sociales cubren algunas de nuestras necesidades en el mundo de la apariencia, como las del reconocimiento, la necesidad de afecto (muchas veces, medida en función de los likes, los retwit, o el número de visualizaciones de vídeos colgados). Las redes te dan la posibilidad de mostrar una imagen de ti que no puedes ser en la vida real e, incluso, el desarrollar distintas identidades, con el peligro que ello supone en una personalidad que se está forjando.
Por otra parte, "las redes no duermen", por lo que algunos jóvenes sufren angustia por no poder responder de inmediato a mensajes durante la noche o durante su asistencia a clases u otras obligaciones, o bien, sí los contestan, a costa de su higiene del sueño y de la repercusión negativa en sus estudios. Por no hablar de lo problemas de conducta en algunos cuando sus padres tratan de poner límites en este sentido.
En 2018, Marina Díaz- Marsá, Presidenta de la Sociedad de Psiquiatría de Madrid, ya declaraba que cada vez hay más diagnósticos en jóvenes depresivos que pasan horas y horas en redes sociales.
En consulta, cada vez más a menudo, además de lo comentado, los profesionales de la salud mental vemos jóvenes víctimas de ciberacoso o de pederastas que contactan con ellos a través de las redes. Los más vulnerables son los/las menores que ya presentan un trastorno de base, como un TDAH o un TAS, de manera que podemos casi hablar de patología dual.
Todo ello hace imprescindible la prevención, por lo que a continuación se explicitan algunas pautas concretas para un buen uso de la redes, principalmente en los más jóvenes:
- Controlar el tiempo de conexión a Internet.
- No creer por completo todo lo que se publica en estos medios.
- Identificar grupos sociales con gustos o intereses similares a los propios.
- No dedicar demasiado tiempo a visualizar publicaciones de otros.
- Evitar pensar de forma irracional, por ejemplo, que es necesario hacer publicaciones que llamen la atención para ser aceptado/a.
- Participar en actividades sociales fuera de la red.
- Realizar actividad física con frecuencia.
- Fomentar otras aficiones y actividades, tanto individuales como en equipo (pero de manera presencial).
- Trabajar la propia autoestima, independiente de la opinión de los demás.
Para concluir, como no podía ser de otra forma, se recomiendas los siguientes enlaces web de cara a la seguridad en Internet:
— Estrategia Nacional de Ciberseguridad 2019. https://www.dsn.gob.es/es/documento/estrategia-nacional-ciberseguridad-2019
.Redes sopciales - RR.SS. - salud mental - ansiedad - depresión - dependencia psicológica - impulsividad - autoestima - higiene del sueño - TDAH - TAS - Quirónsalud - Hospital Universitario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Daniel Martín Fernández Mayoralas - neuropediatra1 comentario - 201916ene
Plagiocefalia y su tratamiento
Tipos de deformidades posturales
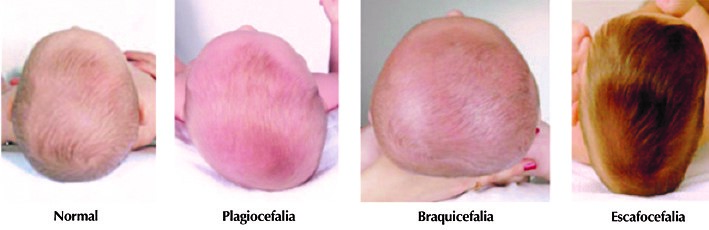
La Plagiocefalia es una malformación asimétrica de la cabeza provocada por presiones externas. Puede ser causada por una serie de factores relacionados con el posicionamiento, tales como un largo periodo de tiempo en la incubadora, el proceso durante el nacimiento o la posición en el útero. Aunque sobre todo es la preferencia del bebé por una determinada postura al dormir o al estar acostado, lo que conduce al problema. Una de las causas frecuentes en este último caso, es una alteración física denominada torticolis (85% de los casos),producida por el espacio reducido del bebé en el útero. Existe una asimetría en los músculos del cuello que flexionan la cabeza y la inclinan hacia el lado afectado girando la cara hacia el lado opuesto. Cuando uno de estos músculos está tenso, obliga al niño a dormir sobre el lado afectado y la parte de atrás de la cabeza tiende a aplanarse debido a la maleabilidad de los huesos craneales de los bebés. Todo ese lado, incluida la oreja, se desplaza hacia adelante y la cabeza adquiere una forma peculiar. En muchos casos, se produce un abombamiento de la frente en ese mismo lado por el mismo motivo.
Existe otra enfermedad denominada Craneosinostosis que provoca una deformación de la cabeza y puede parecerse a la Plagiocefalia posicional, sin embargo, esta alteración es causada por una fusión prematura de una o más suturas del cráneo. Éste puede adoptar una forma inusual si una o más suturas se cierran antes de que el cerebro alcance su pleno desarrollo. Los neuropediatras pueden diferenciar estas dos alteraciones basándose en la historia clínica y en un examen físico, aunque el diagnóstico puntualmente ruede requerir de pruebas radiológicas. Si un bebe padece Craneosinostosis se requiere cirugía para realinear las placas óseas del cráneo y facilitar el crecimiento normal.
La Plagiocefalia posicional no afecta al cerebro y no es causa de retraso mental ni de ningún tipo de alteración neurológica o motora, es meramente estética, salvo que exista algún síndrome asociado a la misma, lo que puede formar parte del diagnóstico diferencial a cargo del neurólogo infantil.
El cráneo de un bebé es fino y maleable y si los bebés pasan un largo periodo de tiempo en una misma posición, la cabeza se puede deformar y hacerse asimétrica.
Si el bebé tiene menos de 3 ó 4 meses y la asimetría es leve se recomienda una postura alternativa a la hora de dormir, con la cabeza hacia el lado contrario.
Existen varios modos de conseguir esto y entre ellos están:
- La estimulación del bebé con juguetes hacia el lado contrario a la deformidad.
- Poner el bebé boca abajo, sobre su barriga, durante el día.
Su doctor también puede recomendarle ejercicios de estiramiento si su hijo sufre torticolis. Si el reposicionamiento y los ejercicios no son efectivos, su médico especialista puede recomendarle la colocación de una ortesis de remodelación craneal (Casco).
Si la Plagiocefalia viene acompañada de Torticolis debe ser tratada a la vez que el tratamiento remodelador ortopédico.
Una detección precoz de la plagiocefalia es clave para el éxito del tratamiento, durante los dos primeros meses de vida se produce la deformidad teniendo su momento álgido el tercer mes, una detección temprana durante esos dos primeros meses puede ayudar a evitar tanto el aumento de la deformidad como su mejora, tanto padres como pediatras son parte importante en esta detección y si la deformidad ya se ha producido derivar al bebé al neuropediatra para descartar una craneosinostosis y si es así iniciar el tratamiento remodelador cuanto antes.
El momento óptimo para el inicio es el período que va desde los 4 a los 8 meses de edad, pasado este periodo el tratamiento remodelador pierde mucha efectividad debido a la disminución del crecimiento del cráneo y la capacidad de direccionar el mismo.
Si durante el tratamiento el especialista considera que la remodelación realizada es óptima, se puede retirar la ortesis sin riesgo de perder lo ya recuperado, pues la deformidad no suele volver a aparecer.
- 201921may
Breve guía del Síndrome de Klinefelter (SK) - Primera parte

Durante las pasadas semanas se han diagnosticado en nuestro servicio algunos niños con Síndrome de Klinefelter (SK). Dada su elevada frecuencia en la población general hemos querido comentar algunos aspectos sobre el mismo que pudiera interesar a bastantes personas.
Descripción.
En el SK existe una duplicación de material genético que afecta a prácticamente la totalidad del cromosoma X. Este síndrome describe un grupo de trastornos cromosómicos en los que hay al menos un cromosoma X extra en un cariotipo masculino (que normalmente es 46, XY), por este motivo el SK ocurre sólo en varones y las manifestaciones se deben a la presencia de un cromosoma X extra (47, XXY). La aneuploidía 47, XXY es el trastorno de los cromosomas sexuales más frecuente en humanos, con una prevalencia de 1/500 varones. El síndrome 48, XXYY, puede ser considerado como una variante del SK, es mucho menos frecuente (1/50.000 varones) y con una gravedad mayor de afectación intelectual y psiquiátrica que los pacientes con SK, especialmente en lo referido a la conducta, siendo habitualmente más explosivos.
Diagnóstico
Normalmente el diagnóstico se realiza tras realizar un cariotipo o unos CG-arrays tras consultar al médico alguno o algunos de los síntomas que se presentan en los siguientes apartados.
Fenotipo (apariencia).
Los pacientes con SK son individuos altos y delgados, con piernas relativamente largas. Los hallazgos más frecuentes en los niños con SK que no han alcanzado la adolescencia son los genitales externos pequeños y las extremidades inferiores largas. Puede existir criptorquidia (descenso incompleto de uno o ambos testículos a través del canal inguinal hacia el escroto). La característica más común del SK es la esterilidad, por ello, durante la pubertad, pueden objetivarse signos de hipogonadismo (los testículos se desarrollan poco y son pequeños y de poca consistencia), así como tendencia al sobrepeso u obesidad. Suelen tener poco vello y acumulan más grasa en glúteos y caderas, desarrollando algunos pacientes ginecomastia (agrandamiento patológico de las glándulas mamarias en el hombre).
Neurodesarrollo.
Lenguaje-lectura.
Entre los problemas del neurodesarrollo más característicos del SK se encuentran los trastornos del habla y del lenguaje: pueden oscilar entre las dislalias (pronunciación), el trastorno específico del lenguaje, como, por ejemplo, la disfasia del desarrollo, o un déficit semántico-pragmático (esto es, dificultades en la comunicación social). En ocasiones existen rasgos de trastorno del espectro autista, aunque el diagnóstico completo de este último es raro. El trastorno del aprendizaje de la lectura, incluyendo la dislexia, también es frecuente, como resultado, al menos en parte, de estas deficiencias. Los pacientes con SK casi nunca tienen discapacidad intelectual global.
Trastornos interiorizantes y relaciones sociales.
Los SK pueden tener dificultades en la socialización (inseguridad, timidez, etcétera), por lo que les puede costar relacionarse adaptativamente con niños y adolescentes de su grupo de edad). Existe una tendencia a padecer trastornos interiorizantes, fundamentalmente ansiedad, pero con menor frecuencia distimia o incluso depresión. El SK suele tender, por lo tanto, al neuroticismo o inestabilidad emocional, rasgo psicológico relativamente estable y que conlleva inestabilidad e inseguridad emocional, tasas elevadas de ansiedad, estado continuo de preocupación y tensión, con tendencia a la culpabilidad y generalmente unido a sintomatología psicosomática (cefaleas, dolor abdominal, etcétera). Algunos pacientes con SK no tienen problemas significativos desde este punto de vista, y es que no hay dos SK iguales.
Trastorno por déficit de atención (TDAH).
Uno de los padecimientos más frecuentes del SK es la disfunción frontal-ejecutiva. Esta disfunción incluye el TDAH, más frecuente en su forma "sin hiperactividad" o de presentación inatenta, que contribuye negativamente a una peor resolución de los problemas del aprendizaje de la lectura y finalmente a la autoestima y al aumento del estrés y la ansiedad. Se ha comprobado la existencia de déficits neuropsicológicos en SK sometidos a tareas de exploración de funciones ejecutivas. Muchas veces, estas dificultades se aprecian más con el aumento de edad y, por ende, de dificultades académicas en función del nivel de estudios que se proponga el paciente. Además, el TDAH puede empeorar las habilidades sociales en individuos que acostumbran a ser tímidos y con frecuencia ansiosos socialmente. Cuando existe un TDAH moderado o grave, diagnosticado a través de la clínica, las escalas y los test neuropsicológicos, el tratamiento con fármacos apropiados es esencial para el futuro del niño con SK.
Psicomotricidad.
Los pacientes con SK también muestran alteraciones en los hitos motores. Típicamente son algo más torpes, especialmente en coordinación motora gruesa, teniendo mejor destreza motora fina. Algunos también pueden mostrar dificultades para la resolución deliberada de problemas. Probablemente estos déficits son independientes del retraso en el desarrollo de otras aptitudes.
Pese a estas aseveraciones existen multitud de niños con SK sin déficit significativo alguno en cuanto al neurodesarrollo, lo que nos da idea del amplio espectro de fenotipos distintos posible en esta cromosomopatía.
La detección precoz del SK, y la consiguiente intervención sobre los errores fonológicos y atencionales-ejecutivos (incluyendo medicación para el TDAH si es necesaria) puede reducir el riesgo de trastorno del lenguaje y lectura posteriores, pudiendo optimizarse las dificultades académicas y, en última instancia, sociales y de comportamiento.
Fertilidad.
El SK es una de las principales causas de azoospermia (ausencia de espermatozoides en el semen) no obstructiva. Aunque en el inicio de la adolescencia, la mayoría de los individuos con SK tienen células germinales en sus testículos, la espermatogonia (células madre especializadas en diferenciarse para dar lugar a los espermatozoides) tienen dificultades para entrar en meiosis y con la edad aumentan la fibrosis y la hialinización (engrosamiento) de los tejidos testiculares. Por este motivo los adultos con SK son considerados tradicionalmente como infértiles.
Síndrome de Klinefelter - SK - cromosoma 47 - cromosoma XXY - aneuploidía - criptorquidia - esterilidad - hipogonadismo - sobre peso - obesidad - ginecomastia - dislaias - disfasia del desarrollo - dislexia - trastornos interiorizantes - ansiedad - distimia - depresión - neuroticismo - TDAH - TDA - azoospermia - neuropediatra - complejo hospitalario Ruber Juan Bravo - Dr. Daniel Martín16 comentarios - 20195jul
Breve guía del Síndrome de Klinefelter (SK) - Segunda parte

Seguimiento.
El seguimiento médico es importante y multidisciplinar desde el inicio del diagnóstico. El médico o pediatra de familia constituye la piedra angular del seguimiento en el SK.
El Neuropediatra debe de interrogar por posibles crisis epilépticas febriles o si hay síntomas de crisis no febriles (por ejemplo, ausencias o crisis parciales complejas). La población XXY tiene una mayor incidencia de crisis epilépticas que la población general, aunque de existir, el control no suele ser muy difícil. Además debe de procurar tratamiento farmacológico para los problemas de atención (TDAH) o de otro tipo de trastorno de conducta cuando sea preciso. Puede y debe recurrir al psiquiatra infantil cuando sea necesario. También ha de estar atento a la posibilidad de que aparezca temblor esencial benigno, relativamente frecuente en el SK, y también tratable.
Inmediata e independientemente de la edad el paciente, éste debe ser evaluado por un endocrinólogo y un urólogo infantil. Con frecuencia hay que tratar la criptorquidia o el hipospadias, si existe, durante los dos primeros años de vida. En ocasiones, si existe micropene, pueden usarse pequeñas dosis de testosterona.
Los neuropediatras (y a veces los psiquiatras infantiles), logopedas, pedagogos y psicólogos son importantes de cara al manejo de las dificultades del neurodesarrollo.
Los traumatólogos deben evaluar la masa muscular, que puede estar poco desarrollada (se cansan con más facilidad), los pies planos, así como la afectación de la articulación del codo, del dedo meñique (clinodactilia), y de la espalda (escoliosis). Se debe promover una dieta adecuada y fomentar ejercicio físico. Los reumatólogos pueden sospechar enfermedades reumáticas, más frecuentes que en la población general.
Los cardiólogos deben realizar al menos un estudio cardiológico, para descartar cualquier defecto de conducción o anomalía cardiaca congénita (son raras, la más frecuente, el prolapso de la válvula mitral).
Tratamiento.
El tratamiento sustitutivo hormonal (habitualmente con derivados de la testosterona) en diferentes etapas (para inducir progresivamente y mantener una virilización apropiada a la edad) ha demostrado aumentar la energía y el rendimiento muscular además de mejorar el humor, concentración y relaciones con los demás. Se suele aconsejar su uso prolongado para la prevención de osteoporosis, obesidad, síndrome metabólico, dislipemia y diabetes.
La monitorización de la terapia incluye analíticas u otras pruebas que determinará el endocrinólogo infantil.
Aunque clásicamente que ha dicho que el tratamiento sustitutivo hormonal debe empezarse cuando se inicia la pubertad, alrededor de los 12 años, parece que su inicio precoz, dictaminado por un endocrinólogo infantil especialista, puede mejorar ciertos aspectos del neurodesarrollo, incluyendo atención y aprendizaje, así como la ansiedad y la timidez secundaria a ésta (ansiedad social). Éste promoverá el desarrollo de los caracteres sexuales secundarios masculinos, el crecimiento testicular (pero no tanto la función) y el aumento de la masa muscular lo que suele implicar un aumento de la autoestima, y suele mejorar la atención y el resto de los síntomas TDAH.
El tratamiento debe ser iniciado y seguido por el endocrinólogo infantil, para individualizar la dosis en cada caso, evaluar el uso de suplementos de calcio/vitamina D y también vigilar la aparición de efectos secundarios, como la hipercolesterolemia o la poliglobulia (que a veces conduce a trombosis). Algunos estudios recientes que han usado alta tecnología consideran que se puede obtener esperma durante la edad pediátrica y tras inyecciones intracitoplasmáticas, conseguir la paternidad en algunos casos (quizás entre el 30-40% de los casos). Suelen llevar a cabo este procedimiento los urólogos especializados. La criopreservación de muestras de semen podría ser ofrecida a los niños antes de iniciar la terapia con testosterona. No hay que olvidar el riesgo de que la paternidad biológica en pacientes con SK puede asociar a fetos con otras cromosomopatías sexuales o de otro tipo. En todo caso, este caso debe ser llevado por un especialista en fertilidad, lo que está muy lejos del alcance de mis conocimientos.
Síndrome de Klinefelter - SK - cromosoma 47 - cromosoma XXY - aneuploidía - criptorquidia - esterilidad - hipogonadismo - sobre peso - obesidad - ginecomastia - dislaias - disfasia del desarrollo - dislexia - trastornos interiorizantes - ansiedad - distimia - depresión - neuroticismo - TDAH - TDA - azoospermia - neuropediatra - complejo hospitalario Ruber Juan Bravo - Dr. Daniel Martín0 comentarios
Sobre este blog
Blog sobre los temas relacionados con la neuropedciatría: déficit de atención, hiperactividad, epilepsia, cefaleas, tics, encefalitis, problemas escolares, etc.

Archivo del blog
 2.024
2.024
- 2.023
- 2.022
- 2.021
- 2.020
- 2.019
- 2.018
- Diciembre
- Noviembre
- Octubre
- Julio
- Mayo
- Abril
- Febrero
- Recomendaciones generales y específicas para el trastorno por déficit de atención/hiperactividad -TDAH- (III). Recomendaciones específicas.
- El Dr. Daniel Martín Fernández-Mayoralas ganador del “Concurso de Casos Clínicos sobre el abordaje farmacológico de pacientes con TDAH" organizado por el Grupo Saned
- Enero
- 2.017
- 2.016
Últimas entradas
- Síndrome cognitivo afectivo del cerebelo: Un diagnóstico a tener en cuenta.
- Síndrome de Pitt-Hopkins
- Tratamiento Cognitivo Conductual en adolescentes con Trastorno por Déficit de Atención con/sin Hiperactividad (TDAH)
- Ejercicio y TDAH
- Empoderando a los Docentes: Estrategias para detectar y abordar dificultades en el aula. (III)
Colaboraciones




La finalidad de este blog es proporcionar información de salud que, en ningún caso sustituye la consulta con su médico. Este blog está sujeto a moderación, de manera que se excluyen de él los comentarios ofensivos, publicitarios, o que no se consideren oportunos en relación con el tema que trata cada uno de los artículos.
Quirónsalud no se hace responsable de los contenidos, opiniones e imágenes que aparezcan en los "blogs". En cualquier caso, si Quirónsalud es informado de que existe cualquier contenido inapropiado o ilícito, procederá a su eliminación de forma inmediata.
Los textos, artículos y contenidos de este BLOG están sujetos y protegidos por derechos de propiedad intelectual e industrial, disponiendo Quirónsalud de los permisos necesarios para la utilización de las imágenes, fotografías, textos, diseños, animaciones y demás contenido o elementos del blog. El acceso y utilización de este Blog no confiere al Visitante ningún tipo de licencia o derecho de uso o explotación alguno, por lo que el uso, reproducción, distribución, comunicación pública, transformación o cualquier otra actividad similar o análoga, queda totalmente prohibida salvo que medie expresa autorización por escrito de Quirónsalud.
Quirónsalud se reserva la facultad de retirar o suspender temporal o definitivamente, en cualquier momento y sin necesidad de aviso previo, el acceso al Blog y/o a los contenidos del mismo a aquellos Visitantes, internautas o usuarios de internet que incumplan lo establecido en el presente Aviso, todo ello sin perjuicio del ejercicio de las acciones contra los mismos que procedan conforme a la Ley y al Derecho.




© 2024 Quirónsalud - Todos los derechos reservados
