Quirónsalud








Blog del Dr. Daniel Martín Fernández-Mayoralas. Neurología. Complejo Hospitalario Ruber Juan Bravo y Hospital Universitario Quirónsalud Madrid
- 202330may
Tratamientos de precisión en epilepsia
Los últimos 30 años han visto la introducción en la práctica clínica de más de 20 fármacos antiepilépticos de segunda generación. A pesar de este armamento farmacológico ampliado, las crisis epilépticas son resistentes al tratamiento en aproximadamente un tercio de las personas con epilepsia, y no se pueden controlar por completo: en el caso de las encefalopatías epilépticas (y del desarrollo) el resultado puede ser peor. Debemos entender que los fármacos anticrisis disponibles se han desarrollado con el objetivo de suprimir los síntomas (crisis), y no fueron diseñados para abordar las etiologías y los mecanismos específicos responsables de la epilepsia.

La medicina de precisión se puede definir como la aplicación de "tratamientos dirigidos a las necesidades de pacientes individuales sobre la base de características genéticas, de biomarcadores, fenotípicas o psicosociales", y tiene como objetivo mejorar los resultados clínicos al adaptar los tratamientos a las necesidades y características de cada individuo.
La mayoría de las epilepsias monogénicas consisten en encefalopatías epilépticas, cada una de las cuales se asocia típicamente con una variedad de tipos de crisis generalizadas y/o focales. Por lo tanto, la mayoría de los tratamientos empíricos utilizados en estas epilepsias implican el uso de fármacos anticrisis de amplio espectro, habiendo muchos ejemplos de este tipo de fármacos.
Los tratamientos de precisión se basan en la comprensión de la función de los genes mutados y la aplicación de intervenciones que se sabe o se espera que corrijan la disfunción causada por los productos del gen mutado. Esto puede ocurrir bajo diferentes escenarios: la selección de fármacos dirigidos específicamente a los mecanismos patogénicos de la enfermedad, el desarrollo de terapias dirigidas basadas en moléculas novedosas, el uso de tratamientos dietéticos o constituyentes alimentarios para corregir defectos metabólicos específicos o la reutilización de medicamentos aprobados originalmente para otras indicaciones.
En el momento actual están comenzando a surgir varios tratamientos de precisión para muchas de estas afecciones, impulsados por los continuos avances en nuestra comprensión de los mecanismos moleculares subyacentes a estas enfermedades. Por este motivo, aunque estos tratamientos no pueden dirigirse con la precisión que quisiéramos a todos los niños, sí a algunos. Por ello quiero a través de este blog dar importancia a la realización de test genéticos adecuados en, si no todos, al menos, en cualquier paciente epiléptico cuyos fármacos no estén controlando la epilepsia a la perfección.
- 202221abr
ERRORES INNATOS DEL METABOLISMO. EPILEPSIAS GENÉTICAS (III).
La epilepsia causada por errores congénitos en el metabolismo (IEM) es rara. Además de epilepsia, rara como fenómeno aislado, suelen hallarse comorbilidades como el rersao del desarrollo, la discapacidad intelectual, problemas neuropsiquiátricos (TDAH, TEA, etc) y, sobre todo, como síntoma guía: regresión del desarrollo. Estos trastornos, a menudo ocurren debido a una deficiencia hereditaria de una enzima o un cofactor que afecta vías metabólicas y bioquímicas específicas. Debido a que algunos IEM responden a terapias específicas, el diagnóstico temprano es crítico. Aunque los IEM puede presentarse a lo largo de la vida, hay varios que se presentan ya en el período neonatal / lactancia y que involucran deficiencias de vitaminas o cofactores que son susceptibles de tratamiento.
Piridoxina
La epilepsia dependiente de piridoxina (PDE) es una epilepsia autosómica recesiva rara causada por mutaciones en el gen antiquitina. Se presume que este IEM afecta la neurotransmisión inhibitoria de GABA y así aumenta la excitabilidad en el SNC. La presentación es el período del recién nacido, hasta los 3 años, el diagnóstico se sospecha basado en la provocación con piridoxina con electroencefalograma (EEG) monitorizado y confirmado con pruebas genéticas y tratamiento mediante la suplementación con piridoxina. Las deficiencias del neurodesarrollo a largo plazo son variables y no están directamente asociadas con el control de las convulsiones. Las mutaciones en el gen de la piridoxina 5’-fosfato oxidasa (PNPO) conducen a niveles bajos de piridoxal 5’-fosfato (PLP), y su presentación difiere de la PDE, con nacimiento prematuro y crisis epilépticas (pueden ocurrir en el útero), hipoglucemia, acidosis láctica y encefalopatía. El tratamiento es con PLP, la forma activada de piridoxina.
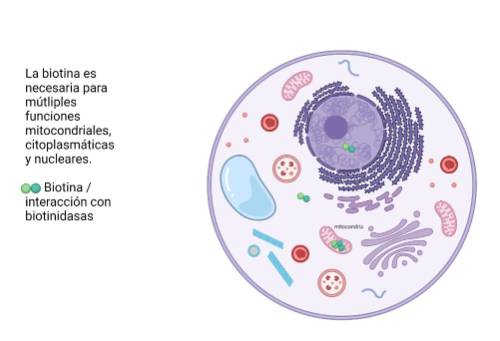
Biotina
Las mutaciones en el gen BTD dan como resultado una deficiencia de biotinidasa, que está involucrada en el reciclaje de la biotina, una enzima necesaria para el metabolismo de la carboxilasa dependiente de biotina. El fenotipo en la deficiencia de biotina incluye crisis epilépticas, retraso marcado en el desarrollo, discapacidad, ataxia, erupción cutánea, alopecia, discapacidad auditiva y visual, e infecciones fúngicas. La deficiencia de biotina se identifica como parte de las pruebas de detección del recién nacido y el tratamiento es con terapia de reemplazo oral de biotina.
Created with BioRender.com
Ácido folínico
Mutaciones del gen FOLR1 que codifica el receptor alfa de folato, un transportador de folato, están asociados con convulsiones sensibles al ácido folínico (FARS) causadas por la interrupción de transporte de folato al SNC. El tratamiento es con ácido folínico pero hay vínculos reportados a PDE, lo que sugiere que el resultado puede mejorar tanto con suplementos de piridoxina como de ácido folínico. La presentación es en la primera infancia con convulsiones, retraso psicomotor e hipotonía.
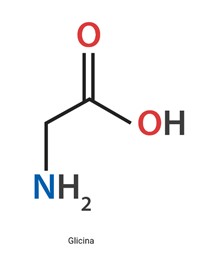
Glicina
Las mutaciones en genes que codifican el sistema enzimático de escisión de glicina, incluido GLDC y AMT, se asocian con hiperglicinemia no cetósica (NKH), que es una enfermedad autosómica recesiva del metabolismo de la glicina. Estas mutaciones provocan acumulación de glicina en el cuerpo con aumentos cuantificables en plasma y líquido cefalorraquídeo (LCR). La presentación clásica, de extrema gravedad, ocurre en el período neonatal con encefalopatía, hipotonía y mioclonías. El patrón de EEG de salvas / supresión indica una gravedad extrema y la epilepsia fármacorresistente con discapacidad intelectual grave y a veces la defunción, es la norma. La intervención es de apoyo dirigido a disminuir los niveles de glicina con benzoato de sodio, pero, en el NKH de inicio neonatal, el pronóstico en cuanto al desarrollo psicomotor sigue siendo muy malo. Las variantes de inicio tardío se consideran formas atenuadas de NKH con un fenotipo menos grave y responden mejor a la terapia con benzoato de sodio y dextrometorfano (antagonista del receptor de N-metil-D-aspartato).
Created with BioRender.com
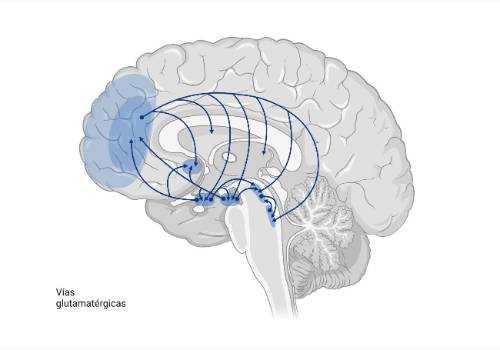
Glutamato
Mutaciones en el gen SLC2A1 que codifica el transportador de glucosa cerebral (GLUT1) conducen al síndrome de la deficiencia de Glut-1, que puede ser por herencia autosómica dominante o por haploinsuficiencia esporádica. El síndrome de deficiencia de GLUT1 se presenta clásicamente con inicio infantil de convulsiones farmacorresistentes, retraso en el desarrollo, microcefalia adquirida, tono y movimientos anormales. Sin embargo, existe un amplio espectro clínico que incluye retraso en el desarrollo, epilepsia, discinesias paroxísticas inducidas por el ejercicio, y deterioro cognitivo. Las convulsiones motoras focales se observan en la infancia y, más tarde, predominan las crisis generalizadas, incluida la epilepsia con ausencias de inicio temprano y la epilepsia mioclónico-astática. Las crisis pueden aumentar antes de las comidas o con el ayuno. Los niveles de glucosa son bajos en LCR (con niveles de glucosa en sangre realizados inmediatamente antes de la punción normales)aunque a veces pueden ser normales en algunos pacientes con deficiencia de GLUT1, por lo que las pruebas genéticas para mutaciones en el gen SLC2A1 son críticas. La dieta cetogénica es una terapia eficaz o la dieta Atkins modificada en casos más leves. Deben evitarse el fenobarbital y el diazepam.
Created by Biorender.com
UNA PERSPECTIVA TERAPÉUTICA.
Las pruebas genéticas se realizan de forma cada vez más rutinaria y precoz en el curso de la epilepsia, por lo que las terapias se van guiando, dentro de lo posible, por estos resultados. Por ejemplo, mutaciones de ganancia de función de los receptores GABA-A que resultan en un tono inhibitorio suprimido pueden no responder bien al tratamiento con un fármaco agonista del receptor GABA que exacerbará la frecuencia de las crisis. El uso de bloqueadores de los canales de sodio tradicionales empeora las crisis del síndrome de Dravet. Otros factores genéticos pueden influir en la respuesta a los fármacos, pero extendería mucho el presente texto. La inmunomodulación se puede usar en epilepsias autoinmunes pero también en epilepsias genéticas. Por ejemplo: síndromes de punta onda durante el sueño (específicamente se ha obtenido respuesta en casos con mutación de GRIN2A). El cannabidiol o, el modulador de la vía de la rapamicina, Everolimus parecen prometedores en casos de Esclerosis Tuberosa (vía mTOR). Se puede considerar la terapia dietética con dieta cetogénica para epilepsias genéticas según el defecto genético subyacente y el síndrome de epilepsia. Un trastorno específico que responde a la dieta cetogénica es el síndrome de deficiencia de GLUT1. Actualmente hay numerosas líneas de investigación sobre las terapias dirigidas a mecanismos genéticos, lo que se denomina terapia génica, pero aún está en proceso preclínico.
 epilepsia - vías metabólicas - bioquímicas específicas - errores congénitos - piridoxina - biotina - ácido folínico - glicina - glutamato - pruebas genéticas - terapias - quirónsalud - Complejo Hospitalario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Danierl Martín - Fernández-Mayoralas0 comentarios
epilepsia - vías metabólicas - bioquímicas específicas - errores congénitos - piridoxina - biotina - ácido folínico - glicina - glutamato - pruebas genéticas - terapias - quirónsalud - Complejo Hospitalario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Danierl Martín - Fernández-Mayoralas0 comentarios - 202214ene
CANALES DE IONES Y EPILEPSIA. EPILEPSIAS GENÉTICAS (I).
En ausencia de una causa adquirida, la presencia de crisis epilépticas recurrentes espontáneas o epilepsia de causa desconocida suscita una fuerte sospecha de una anomalía genética subyacente. Se ha propuesto que las causas genéticas explican para al menos el 30% de las epilepsias1. Las mutaciones genéticas con un fenotipo primario de epilepsia incluyen aquellas que involucran iones canales, proteínas sinápticas y de señalización, y otras moléculas. Con una rápida evolución sofisticación en las pruebas genéticas y una mayor disponibilidad, la lista de anomalías genéticas con un fenotipo de epilepsia primaria está creciendo.
 CANALES DE IONES
CANALES DE IONESCanales de sodio.
La mayoría de las epilepsias genéticas están asociadas con mutaciones en genes que codifican subunidades de Canales iónicos activados por voltaje, particularmente en canales de sodio activados por voltaje, fundamentales para el inicio y la propagación de los potenciales de acción y su localización es crucial para una función neuronal óptima2. Por ejemplo, las mutaciones en el gen SCN1A que codifica la subunidad a Nav1.1 pueden producir un síndrome de Dravet (el 80% de éstos), que cursa con epilepsia, a menudo grave y discapacidad cognitiva. Mutaciones en otros genes del sodio (SCN2A, SCN3A, SCN8A, SCN9A, etcétera) producen síndromes epilépticos de diversa índole. Curiosamente, las crisis epilépticas en pacientes con algunos tipos de mutaciones en el gen SCN2A, a menudo desaparecen a los 18-24 meses de edad3.
Canales de potasio.
El deterioro de las funciones repolarizantes de los canales de potasio puede conducir a una descarga neuronal hiperexcitable y sincrónica. Se han identificado mutaciones de los genes de los canales de potasio en la epilepsia, que incluyen KCNA2, KCNB1 y muchos otros. Por ejemplo, las variantes del gen KCNA2 que codifican Kv1.2 pueden causar encefalopatía epiléptica con ataxia y temblores4. La gravedad de los síndromes epilépticos es extremadamente variables. Las mutaciones en los canales Kv7.2 codificados por KCNQ2 se pueden asociar con convulsiones neonatales familiares o con encefalopatías epilépticas graves, según la mutación que albergue. Otros genes parecen menos variables a su afectación, como el síndrome de Temple Baraitser, por mutaciones en KCNH15,6.
Otros canales.
Otros canales importantes son los canales del cloro. Ciertas variantes de CLCN2 se asocian con mutaciones de SCN1A en pacientes con síndrome de Dravet y encefalopatía epiléptica infantil temprana, lo que sugiere un posible papel de CLCN2 como modificador de genes7. El cotransportador de cloruro de potasio (SLC12A5), puede producir una encefalopatía epiléptica de la primera infancia con convulsiones migratorias focales o incluso una epilepsia generalizada idiopática en mutaciones de SLC12A58. Los canales de calcio dependientes de voltaje conducen una corriente de calcio hacia el interior de la célula y contribuyen a una mayor excitabilidad neuronal. Variantes del gen CACNA1A pueden conducir a una función alterada de Cav2.1 que se asocie con una epilepsia con ausencias leve hasta una encefalopatía devastadora con accidentes cerebrovasculares isquémicos en la lactancia9.
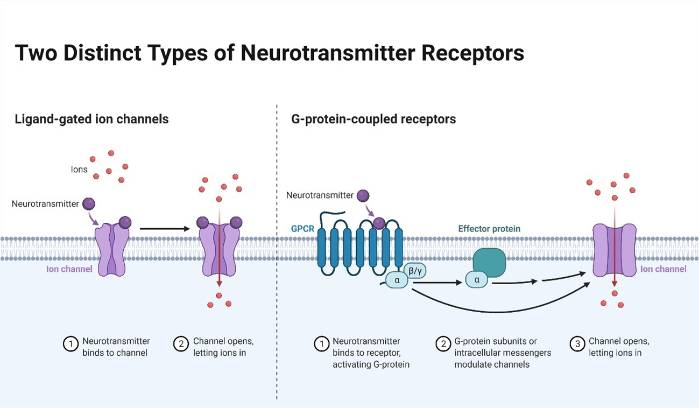
En el otro capítulo nos proponemos hablar de los receptores metabotrópicos (que modulan la respuesta neuronal a muchos sistemas de neurotransmisores y no son canales iónicos "directos" como los receptores de ácido gamma-aminobutírico, los receptores nicotínicos de acetilcolina o los de glutamato, así como de genes que codifican proteínas sinápticas, encargadas de formar y mantener la sinapsis adecuadamente como SYNGAP1 o STXBP1.
 0 comentarios
0 comentarios - 202121dic
Epilepsia: ¿Es importante realizar test genéticos?
El conocimiento de los mecanismos genéticos de la epilepsia ha evolucionado velozmente, especialmente en el último lustro. El abaratamiento de los test genéticos realmente útiles para el diagnóstico causal de las epilepsias, y su clara utilidad e importante rendimiento, está suponiendo una evolución exponencial en la detección de las causas subyacentes a muchos tipos de epilepsia y por ende, una mejoría en el tratamiento de las mismas a corto plazo.
Cada vez hay pacientes con un diagnóstico genético preciso de su epilepsia. La capacidad para identificar una causa genética, de hecho, es tan extraordinaria, que ha superado en ocasiones la capacidad para dirigir un tratamiento específico, pero poco a poco la lista de trastornos que recibirán una terapéutica específica continuará creciendo gracias a la precisión del diagnóstico.
Muchos padres preguntan en consulta: ¿Y de qué sirve saber la causa exacta por la que mi hijo convulsiona’ ¿Se puede hacer algo? La respuesta es que "tal vez" y que es tal vez se irá acercando en los años venideros al "sí".
¿Por qué "tal vez"? Porque a pesar de la explosión del descubrimiento de un gran número de genes asociados con la epilepsia, buena parte de ellas permanece sin resolver (incluidas muchas de las epilepsias más frecuentes, en parte, porque suelen ser más benignas y menos propensas a recibir fondos de investigación). Sin embargo, la investigación del papel de los factores multifactoriales o poligenéticos y la epigenética, así como una mayor comprensión de la variedad de manifestaciones clínicas y el papel de los genes de la epilepsia, a medida que vayamos conociendo más y más, resultará en una medicina de precisión, donde el diagnóstico genético se puede utilizar para elegir el tratamiento más adecuado, o, quien sabe, curar la epilepsia del paciente investigado.
El uso clínico de las pruebas genéticas puede llevar a los pacientes y sus familias al final de una «odisea diagnóstica» mediante la identificación de un diagnóstico preciso, a veces con el potencial de una terapia dirigida o racional para las crisis y las enfermedades asociadas conocidas.
No podemos obviar que YA podemos realizar un tratamiento relativamente específico cuando detectamos una mutación causal en una epilepsia. Algunos ejemplos: Evitar determinados fármacos en determinadas mutaciones genéticas. Usar la dieta cetógena como una opción primaria en ciertas causas de epilepsia. Evitar o, al revés, promover un fármaco según el hallazgo, como es el caso de la carbamazepina o la fenitoína, según el hallazgo encontrado. Usar sustancias poco habituales en el manejo estándar de los síndromes epilépticos como el Inositol, la Piridoxina, el Piridoxal-5-fosfato, la retigabina, la quinidina, la memantina, el everólimus o la uridina, cuando el descubrimiento del gen implicado en la epilepsia nos "dicta" el uso de tales tratamientos infrecuentes.
Solo por esto, aunque actualmente, el conocimiento de la causa exacta de un síndrome epiléptico, no garantice de cura, o incluso, una mejoría (¿Acaso alguna prueba diagnóstica "clásica" garantiza tal cosa?), merece la pena saber a qué se debe exactamente la epilepsia que padece nuestro hijo, porque, quien sabe, quizás esté en el grupo del tratamiento dirigido, o pueda estarlo dentro de muy poco tiempo. SI no se mira, no se sabe, si no se sabe, perdemos una oportunidad.
- 202015ene
TEA: diagnóstico diferencial (II parte). TEA vs otros trastornos

A continuación vamos a exponer algunas características que se dan en una selección de trastornos y cuáles de éstas pueden ayudar a distinguirlos del TEA:
- Síndrome Alcohólico Fetal (SAF). Rasgos faciales característicos (no siempre presentes): fisuras palpebrales cortas, filtro liso, etc… MUY RARAMENTE PRESENTAN UN TEA FLORIDO Y COMPLETO.
- Trastorno del vínculo o del apego.
- Historial de negligencia grave o problemas de salud mental en el cuidador o cuidadores.
- Los déficits sociales tienden a mejorar con el cuidado apropiado.
- Retraso del desarrollo global/discapacidad intelectual. La capacidad de respuesta social y la comunicación social son adecuadas para el nivel de desarrollo.
- Superdotación intelectual.
- Habilidades del lenguaje pragmático normal.
- Los intereses intensos son funcionales, variados, y el paciente es capaz de explicarlos.
- La interacción social generalmente se disfruta.
- Trastorno de la comunicación social. Ausencia de patrones restringidos y repetitivos de comportamiento, intereses o actividades.
- Retraso del desarrollo del lenguaje.
- Interacciones sociales recíprocas normales.
- Existe un deseo e intención de comunicarse normal.
- Juego imaginativo apropiado.
- Trastornos específicos del lenguaje (TEL).
- Interacciones sociales recíprocas normales (no siempre).
- Existe un deseo e intención de comunicarse normal (a veces se aíslan).
- Deseo de comunicación (incluso si falta la competencia, salvo el punto previo).
- Juego imaginativo apropiado (ojo a la edad).
Los retrasos y sobre todo los trastornos específicos del lenguaje (TEL) tienen un impacto en la socialización y pueden confundirse con un TEA. La distinción es particularmente difícil en niños de preescolar. A veces imposible. Sin embargo, existen tres comportamientos que tienen una mayor consistencia en diferenciar TEA de TEL entre los 20 y los 42 meses de edad:
- Señalar lo que le interesa al niño.
- Uso de gestos convencionales.
- Ausencia de comportamientos estereotipados y repetitivos significativos.
A pesar de que estos pacientes pueden mostrar comportamientos de aislamiento, su buena comprensión no verbal y su empatía ayuda a diferenciarlos del TEA.
- Trastorno de aprendizaje no verbal (TANV). Diagnóstico controvertido no incluido como categoría diagnóstica en el manual DSM.
- Deterioro en habilidades sociales y lenguaje pragmático más suave que en TEA.
- Falta de patrones restringidos y repetitivos de comportamiento, intereses u ocupaciones.
- Hipoacusia.
- Interacciones sociales recíprocas normales.
- Buen contacto ocular.
- Las expresiones faciales indican intención de comunicar.
- Epilepsias con punta onda durante el sueño. Por ejemplo: síndrome de Landau-Kleffner.
- Habitualmente tienen desarrollo típico hasta aproximadamente 3 a 6 años de edad.
- Típicamente se presenta con agnosia verbal auditiva (comportándose como si estuviera sordo)
- Síndrome de Rett.
- Casi exclusivo del sexo femenino.
- Desaceleración del crecimiento de la cabeza durante la lactancia.
- Movimientos estereotipados de las manos en línea media.
- Anomalías de la marcha según avanza la enfermedad.
- Patrón respiratorio anormal.
- Trastorno de ansiedad (incluye Ansiedad social y selectiva. Mutismo).
- Comportamiento social no verbal normal y juego imaginativo.
- Falta de intereses circunscritos.
- Ausencia de patrones restringidos y repetitivos de comportamiento, intereses o actividades.
- Trastorno obsesivo compulsivo (TOC).
- Habilidades sociales básicamente normales.
- Lenguaje pragmático normal.
- Los síntomas son una fuente de ansiedad en lugar de un placer.
- Tics y síndrome de Tourette.
- Habilidades sociales normales.
- Lenguaje pragmático normal.
- Pueden asociar TOC.
- Trastorno por estereotipias.
- Habilidades sociales normales.
- Lenguaje pragmático normal.
Trastorno del espectro autista - TEA - Síndrome Alcohólico Fetal - SAF - Trastorno del vínculo o del apego - retraso del desarrollo global - discapacidad intelectual - superdotación intelectual - trastorno de la comunicación social - retraso del desarrollo del lenguaje - trastornos específicos del lenguaje - TEL - trastorno de aprendizaje no verbal - TANV - Hipoacusia - epilepsia - sñindrome de Landau-Klefner - síndrome de Rett - trastorno de ansiedad - trastorno obsesivo compulsivo - TOC - tics - síndrome de Tourette - trastorno por estereotipias - Quirónsalud - complejo hospitalario Ruber Juan Bravo - Dr. Daniel Martín0 comentarios
Sobre este blog
Blog sobre los temas relacionados con la neuropedciatría: déficit de atención, hiperactividad, epilepsia, cefaleas, tics, encefalitis, problemas escolares, etc.

Archivo del blog
 2.024
2.024
- 2.023
- 2.022
- 2.021
- 2.020
- 2.019
- 2.018
- Diciembre
- Noviembre
- Octubre
- Julio
- Mayo
- Abril
- Febrero
- Recomendaciones generales y específicas para el trastorno por déficit de atención/hiperactividad -TDAH- (III). Recomendaciones específicas.
- El Dr. Daniel Martín Fernández-Mayoralas ganador del “Concurso de Casos Clínicos sobre el abordaje farmacológico de pacientes con TDAH" organizado por el Grupo Saned
- Enero
- 2.017
- 2.016
Últimas entradas
- Síndrome cognitivo afectivo del cerebelo: Un diagnóstico a tener en cuenta.
- Síndrome de Pitt-Hopkins
- Tratamiento Cognitivo Conductual en adolescentes con Trastorno por Déficit de Atención con/sin Hiperactividad (TDAH)
- Ejercicio y TDAH
- Empoderando a los Docentes: Estrategias para detectar y abordar dificultades en el aula. (III)
Colaboraciones




La finalidad de este blog es proporcionar información de salud que, en ningún caso sustituye la consulta con su médico. Este blog está sujeto a moderación, de manera que se excluyen de él los comentarios ofensivos, publicitarios, o que no se consideren oportunos en relación con el tema que trata cada uno de los artículos.
Quirónsalud no se hace responsable de los contenidos, opiniones e imágenes que aparezcan en los "blogs". En cualquier caso, si Quirónsalud es informado de que existe cualquier contenido inapropiado o ilícito, procederá a su eliminación de forma inmediata.
Los textos, artículos y contenidos de este BLOG están sujetos y protegidos por derechos de propiedad intelectual e industrial, disponiendo Quirónsalud de los permisos necesarios para la utilización de las imágenes, fotografías, textos, diseños, animaciones y demás contenido o elementos del blog. El acceso y utilización de este Blog no confiere al Visitante ningún tipo de licencia o derecho de uso o explotación alguno, por lo que el uso, reproducción, distribución, comunicación pública, transformación o cualquier otra actividad similar o análoga, queda totalmente prohibida salvo que medie expresa autorización por escrito de Quirónsalud.
Quirónsalud se reserva la facultad de retirar o suspender temporal o definitivamente, en cualquier momento y sin necesidad de aviso previo, el acceso al Blog y/o a los contenidos del mismo a aquellos Visitantes, internautas o usuarios de internet que incumplan lo establecido en el presente Aviso, todo ello sin perjuicio del ejercicio de las acciones contra los mismos que procedan conforme a la Ley y al Derecho.




© 2024 Quirónsalud - Todos los derechos reservados
