Quirónsalud








Blog del Dr. Daniel Martín Fernández-Mayoralas. Neurología. Complejo Hospitalario Ruber Juan Bravo y Hospital Universitario Quirónsalud Madrid
- 202227abr
El síndrome de kabuki (KS)
El síndrome de Kabuki (KS), es una patología con múltiples anomalías. Las más frecuentes: características faciales peculiares que remedan el maquillaje Kabuki -fisuras palpebrales largas y eversión de los párpados, que consiste en que éstos se ven como si se les hubiera dado la vuelta y se observa la parte interior, es decir, la parte que está en contacto con el ojo-, anomalías esqueléticas, engrosamiento de las yemas de los dedos y talla baja, asociadas a discapacidad intelectual. Pese a que se pensaba que era un síndrome muy raro, actualmente sabemos que afecta a cerca de una cada 30.000 personas aproximadamente, por lo que es una causa relativamente común de discapacidad intelectual, a tener siempre en cuenta, de ahí el interés del presente post.
El gen KMT2D, también conocido como MLL2, proporciona instrucciones para producir una enzima llamada metiltransferasa 2D específica de la lisina, que se encuentra en muchos órganos y tejidos del cuerpo. Esta enzima funciona modificando las histonas, agregándolas un grupo metilo (metilación), de este modo, las histonas metiltransferasas controlan la actividad de ciertos genes a nivel de empaquetamiento de la cromatina (la forma en la que se presenta el ADN en el núcleo celular). Así, la enzima codificada por KMT2D parece activar ciertos genes que son importantes para el desarrollo. Se han identificado diversas y múltiples mutaciones en el gen KMT2D, en personas con KS. El KDM6A, ligado al cromosoma X, puede ser responsable con menor frecuencia del KS, siendo más grave en varones. Existe la posibilidad de realizar el diagnóstico genético de esté síndrome en la práctica clínica, a través de una técnica denominada exoma.
Las anomalías estructurales en el KS pueden incluir lo siguiente:
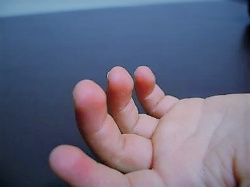
- Yemas de los dedos fetales persistentes (engrosamiento de las yemas de los dedos de las manos); se consideran una de las cinco manifestaciones cardinales del KS y, por lo tanto, se encuentran en una gran proporción de las personas afectadas.
- Oftalmológicas, incluyendo ptosis y estrabismo. Curiosamente, como resultado de la eversión del párpado inferior, los niños con KS pueden mostrar un lagrimeo excesivo, que generalmente no es un problema importante.
- Otológicas (una pista de diagnóstico potencialmente útil es que la mayoría de las personas con KS tienen orejas prominentes y en forma de copa. Los hoyos en el trago y en la región posterior de los pabellones auriculares también son relativamente comunes). La sordera neurosensorial es rara, aunque las otitis medias son comunes y a veces pueden producir pérdida de audición.
- Bucodentales: Labio y/o paladar hendido (un tercio de los niños). Anomalías dentales que incluyen dientes muy separados e hipodoncia (lo que significa que puede haber a veces incisivos superiores laterales ausentes, o incisivos inferiores ausentes, o molares superiores ectópicos y/o segundos premolares faltantes).
- Defectos cardíacos congénitos (cerca de la mitad de los casos, el más frecuente la coartación de aorta).
- Gastrointestinales, incluida la atresia anal. Las más frecuentes están relacionadas con hipotonía, mala coordinación oromotora y dificultades para tragar.
- Genitourinarias, incluyendo criptorquidia (testículo no descendido) en varones.
- Endocrinológicas, incluyendo telarquia prematura (aparición del botón mamario por primera vez en la mujer). En la adolescencia y la edad adulta, más de la mitad de las personas con KS desarrollan obesidad. La deficiencia del crecimiento posnatal es evidente a los 12 meses de edad. La falta de un crecimiento acelerado típico durante la pubertad exacerba la baja estatura. Pueden responder a tratamiento con hormona de crecimiento (GH). Otros hallazgos: hiperinsulinismo, insuficiencia suprarrenal, deficiencia combinada de hormona pituitaria, diabetes insípida, deficiencia franca de hormona de crecimiento, hipotiroidismo, disfunción ovárica primaria, verdadera pubertad precoz.
- Aumento susceptibilidad a enfermedades autoinmunes. No hay evidencia de aumento de cáncer.
- Neurológicas:
- Hipotonía, aunque la hiperlaxitud articular es la regla, lo que afecta al tono pasivo.
- Luxaciones o subluxaciones articulares, que afectan especialmente a las caderas, las rótulas y los hombros. No son infrecuentes, pero como en la mayoría de las condiciones con laxitud articular, este hallazgo mejora con la edad.
- Epilepsia o crisis epilépticas. No suelen ser de difícil control.
- Discapacidad intelectual, generalmente en el rango leve a moderado, en la mayoría de las personas; sin embargo, se han publicado informes de individuos con variantes patogénicas en KMT2D o KDM6A que tienen niveles de coeficiente intelectual superiores a 70. La mayoría de las personas con KS pueden hablar y caminar adecuadamente. No se ha identificado ningún perfil lingüístico específico. Sin embargo, todos los subdominios del lenguaje, incluidos la sintaxis, la morfología, la pragmática y la semántica, pueden verse afectados. En las pruebas neuropsiquiátricas formales, las personas con KS tienden a obtener mejores puntuaciones en las áreas de comprensión de vocabulario y memoria de trabajo y más bajas en las áreas de razonamiento no verbal y velocidad de procesamiento. Las personas con KS tienden a ser descritas como agradables y extrovertidas. En un subconjunto de individuos afectados existe TDAH. Rara vez se han informado otros trastornos: ansiedad, trastornos del espectro autista, trastornos de conducta, trastornos del sueño, etcétera.
El médico especialista, la mayor parte de las veces el Neuropediatra, debe decidir las pruebas a realizar en cada caso (ecografía renal/abdominal, EEG, interconsulta a cardiología, etc.) según los síntomas y signos detectados y/o esperados en cada paciente, de forma individualizada.
BIBLIOGRAFÍA:
Adam MP et al. Kabuki Syndrome. 2011 Sep 1 [Updated 2021 Jul 15]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022.
Banka S et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. European Journal of Human Genetics (2012) 20, 381–388.
Lepri FR. Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature. Int. J. Mol. Sci. 2018, 19, 82; doi:10.3390/ijms19010082.
- 202221abr
ERRORES INNATOS DEL METABOLISMO. EPILEPSIAS GENÉTICAS (III).
La epilepsia causada por errores congénitos en el metabolismo (IEM) es rara. Además de epilepsia, rara como fenómeno aislado, suelen hallarse comorbilidades como el rersao del desarrollo, la discapacidad intelectual, problemas neuropsiquiátricos (TDAH, TEA, etc) y, sobre todo, como síntoma guía: regresión del desarrollo. Estos trastornos, a menudo ocurren debido a una deficiencia hereditaria de una enzima o un cofactor que afecta vías metabólicas y bioquímicas específicas. Debido a que algunos IEM responden a terapias específicas, el diagnóstico temprano es crítico. Aunque los IEM puede presentarse a lo largo de la vida, hay varios que se presentan ya en el período neonatal / lactancia y que involucran deficiencias de vitaminas o cofactores que son susceptibles de tratamiento.
Piridoxina
La epilepsia dependiente de piridoxina (PDE) es una epilepsia autosómica recesiva rara causada por mutaciones en el gen antiquitina. Se presume que este IEM afecta la neurotransmisión inhibitoria de GABA y así aumenta la excitabilidad en el SNC. La presentación es el período del recién nacido, hasta los 3 años, el diagnóstico se sospecha basado en la provocación con piridoxina con electroencefalograma (EEG) monitorizado y confirmado con pruebas genéticas y tratamiento mediante la suplementación con piridoxina. Las deficiencias del neurodesarrollo a largo plazo son variables y no están directamente asociadas con el control de las convulsiones. Las mutaciones en el gen de la piridoxina 5’-fosfato oxidasa (PNPO) conducen a niveles bajos de piridoxal 5’-fosfato (PLP), y su presentación difiere de la PDE, con nacimiento prematuro y crisis epilépticas (pueden ocurrir en el útero), hipoglucemia, acidosis láctica y encefalopatía. El tratamiento es con PLP, la forma activada de piridoxina.
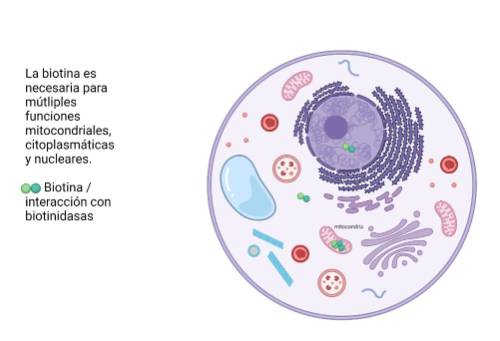
Biotina
Las mutaciones en el gen BTD dan como resultado una deficiencia de biotinidasa, que está involucrada en el reciclaje de la biotina, una enzima necesaria para el metabolismo de la carboxilasa dependiente de biotina. El fenotipo en la deficiencia de biotina incluye crisis epilépticas, retraso marcado en el desarrollo, discapacidad, ataxia, erupción cutánea, alopecia, discapacidad auditiva y visual, e infecciones fúngicas. La deficiencia de biotina se identifica como parte de las pruebas de detección del recién nacido y el tratamiento es con terapia de reemplazo oral de biotina.
Created with BioRender.com
Ácido folínico
Mutaciones del gen FOLR1 que codifica el receptor alfa de folato, un transportador de folato, están asociados con convulsiones sensibles al ácido folínico (FARS) causadas por la interrupción de transporte de folato al SNC. El tratamiento es con ácido folínico pero hay vínculos reportados a PDE, lo que sugiere que el resultado puede mejorar tanto con suplementos de piridoxina como de ácido folínico. La presentación es en la primera infancia con convulsiones, retraso psicomotor e hipotonía.
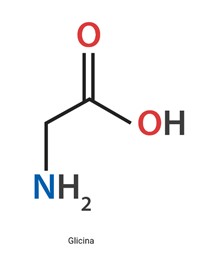
Glicina
Las mutaciones en genes que codifican el sistema enzimático de escisión de glicina, incluido GLDC y AMT, se asocian con hiperglicinemia no cetósica (NKH), que es una enfermedad autosómica recesiva del metabolismo de la glicina. Estas mutaciones provocan acumulación de glicina en el cuerpo con aumentos cuantificables en plasma y líquido cefalorraquídeo (LCR). La presentación clásica, de extrema gravedad, ocurre en el período neonatal con encefalopatía, hipotonía y mioclonías. El patrón de EEG de salvas / supresión indica una gravedad extrema y la epilepsia fármacorresistente con discapacidad intelectual grave y a veces la defunción, es la norma. La intervención es de apoyo dirigido a disminuir los niveles de glicina con benzoato de sodio, pero, en el NKH de inicio neonatal, el pronóstico en cuanto al desarrollo psicomotor sigue siendo muy malo. Las variantes de inicio tardío se consideran formas atenuadas de NKH con un fenotipo menos grave y responden mejor a la terapia con benzoato de sodio y dextrometorfano (antagonista del receptor de N-metil-D-aspartato).
Created with BioRender.com
Glutamato
Mutaciones en el gen SLC2A1 que codifica el transportador de glucosa cerebral (GLUT1) conducen al síndrome de la deficiencia de Glut-1, que puede ser por herencia autosómica dominante o por haploinsuficiencia esporádica. El síndrome de deficiencia de GLUT1 se presenta clásicamente con inicio infantil de convulsiones farmacorresistentes, retraso en el desarrollo, microcefalia adquirida, tono y movimientos anormales. Sin embargo, existe un amplio espectro clínico que incluye retraso en el desarrollo, epilepsia, discinesias paroxísticas inducidas por el ejercicio, y deterioro cognitivo. Las convulsiones motoras focales se observan en la infancia y, más tarde, predominan las crisis generalizadas, incluida la epilepsia con ausencias de inicio temprano y la epilepsia mioclónico-astática. Las crisis pueden aumentar antes de las comidas o con el ayuno. Los niveles de glucosa son bajos en LCR (con niveles de glucosa en sangre realizados inmediatamente antes de la punción normales)aunque a veces pueden ser normales en algunos pacientes con deficiencia de GLUT1, por lo que las pruebas genéticas para mutaciones en el gen SLC2A1 son críticas. La dieta cetogénica es una terapia eficaz o la dieta Atkins modificada en casos más leves. Deben evitarse el fenobarbital y el diazepam.
Created by Biorender.com
UNA PERSPECTIVA TERAPÉUTICA.
Las pruebas genéticas se realizan de forma cada vez más rutinaria y precoz en el curso de la epilepsia, por lo que las terapias se van guiando, dentro de lo posible, por estos resultados. Por ejemplo, mutaciones de ganancia de función de los receptores GABA-A que resultan en un tono inhibitorio suprimido pueden no responder bien al tratamiento con un fármaco agonista del receptor GABA que exacerbará la frecuencia de las crisis. El uso de bloqueadores de los canales de sodio tradicionales empeora las crisis del síndrome de Dravet. Otros factores genéticos pueden influir en la respuesta a los fármacos, pero extendería mucho el presente texto. La inmunomodulación se puede usar en epilepsias autoinmunes pero también en epilepsias genéticas. Por ejemplo: síndromes de punta onda durante el sueño (específicamente se ha obtenido respuesta en casos con mutación de GRIN2A). El cannabidiol o, el modulador de la vía de la rapamicina, Everolimus parecen prometedores en casos de Esclerosis Tuberosa (vía mTOR). Se puede considerar la terapia dietética con dieta cetogénica para epilepsias genéticas según el defecto genético subyacente y el síndrome de epilepsia. Un trastorno específico que responde a la dieta cetogénica es el síndrome de deficiencia de GLUT1. Actualmente hay numerosas líneas de investigación sobre las terapias dirigidas a mecanismos genéticos, lo que se denomina terapia génica, pero aún está en proceso preclínico.
 epilepsia - vías metabólicas - bioquímicas específicas - errores congénitos - piridoxina - biotina - ácido folínico - glicina - glutamato - pruebas genéticas - terapias - quirónsalud - Complejo Hospitalario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Danierl Martín - Fernández-Mayoralas0 comentarios
epilepsia - vías metabólicas - bioquímicas específicas - errores congénitos - piridoxina - biotina - ácido folínico - glicina - glutamato - pruebas genéticas - terapias - quirónsalud - Complejo Hospitalario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Danierl Martín - Fernández-Mayoralas0 comentarios
Sobre este blog
Blog sobre los temas relacionados con la neuropedciatría: déficit de atención, hiperactividad, epilepsia, cefaleas, tics, encefalitis, problemas escolares, etc.

Archivo del blog
 2.024
2.024
- 2.023
- 2.022
- 2.021
- 2.020
- 2.019
- 2.018
- Diciembre
- Noviembre
- Octubre
- Julio
- Mayo
- Abril
- Febrero
- Recomendaciones generales y específicas para el trastorno por déficit de atención/hiperactividad -TDAH- (III). Recomendaciones específicas.
- El Dr. Daniel Martín Fernández-Mayoralas ganador del “Concurso de Casos Clínicos sobre el abordaje farmacológico de pacientes con TDAH" organizado por el Grupo Saned
- Enero
- 2.017
- 2.016
Últimas entradas
- Síndrome cognitivo afectivo del cerebelo: Un diagnóstico a tener en cuenta.
- Síndrome de Pitt-Hopkins
- Tratamiento Cognitivo Conductual en adolescentes con Trastorno por Déficit de Atención con/sin Hiperactividad (TDAH)
- Ejercicio y TDAH
- Empoderando a los Docentes: Estrategias para detectar y abordar dificultades en el aula. (III)
Colaboraciones




La finalidad de este blog es proporcionar información de salud que, en ningún caso sustituye la consulta con su médico. Este blog está sujeto a moderación, de manera que se excluyen de él los comentarios ofensivos, publicitarios, o que no se consideren oportunos en relación con el tema que trata cada uno de los artículos.
Quirónsalud no se hace responsable de los contenidos, opiniones e imágenes que aparezcan en los "blogs". En cualquier caso, si Quirónsalud es informado de que existe cualquier contenido inapropiado o ilícito, procederá a su eliminación de forma inmediata.
Los textos, artículos y contenidos de este BLOG están sujetos y protegidos por derechos de propiedad intelectual e industrial, disponiendo Quirónsalud de los permisos necesarios para la utilización de las imágenes, fotografías, textos, diseños, animaciones y demás contenido o elementos del blog. El acceso y utilización de este Blog no confiere al Visitante ningún tipo de licencia o derecho de uso o explotación alguno, por lo que el uso, reproducción, distribución, comunicación pública, transformación o cualquier otra actividad similar o análoga, queda totalmente prohibida salvo que medie expresa autorización por escrito de Quirónsalud.
Quirónsalud se reserva la facultad de retirar o suspender temporal o definitivamente, en cualquier momento y sin necesidad de aviso previo, el acceso al Blog y/o a los contenidos del mismo a aquellos Visitantes, internautas o usuarios de internet que incumplan lo establecido en el presente Aviso, todo ello sin perjuicio del ejercicio de las acciones contra los mismos que procedan conforme a la Ley y al Derecho.




© 2024 Quirónsalud - Todos los derechos reservados
