Quirónsalud







Blog del Dr. Daniel Martín Fernández-Mayoralas. Neurología. Complejo Hospitalario Ruber Juan Bravo y Hospital Universitario Quirónsalud Madrid

- 20213mar
Ortesis craneales
A menudo los pacientes nos preguntan sobre el mecanismo de funcionamiento de las ortesis craneales cuando existe plagiocefalia, braquicefalia o dolicocefalia.
Para las cuestiones básicos recomendamos acudir al primer capítulo sobre este tema, https://www.quironsalud.es/blogs/es/neuropediatra/plagiocefalia-tratamiento
 .
.Los principios de la intervención mediante ortesis para la plagiocefalia en general se basan en proporcionar un contacto total en las áreas en las cuales se pretende frenar el crecimiento del cráneo para mejorar la deformidad. Se debe permitir un espacio entre la ortesis y el cráneo en las áreas donde se desea que la cabeza crezca. Existe una ventana de oportunidad crítica entre los tres y los nueve meses de edad, en algunos casos hasta los 12 meses de edad cuando la cabeza se está formando activamente. El casco, esto es la ortesis, siguiendo estas pautas va a conseguir que crecimiento cefálico se produzca con normalidad aliviando la deformidad.



- 202120ene
Retraso psicomotor en la infancia (IV Parte): evaluación del desarrollo psicomotor

Debido a la complejidad de este tema, vamos a describir sucintamente las posibles pruebas con las que los padres pueden encontrarse cuando acuden al Neuropediatra. NO hay ninguna obligatoria y otras que no aparecen, para ser lo más concretos posibles.
- Estudios analíticos: deberá valorarse la realización de una analítica con diferentes parámetros que determinará el médico. Ante la presencia de signos que apunten hacia un posible origen metabólico, el examen analítico será ampliado en consecuencia, a veces con determinación de parámetros no sanguíneos como orina o líquido cefalorraquídeo.
- Estudios neurorradiológicos: la utilidad de los estudios mediante resonancia magnética cerebral (RM) dependerá de los hallazgos clínicos y la severidad del retraso.
- Estudios genéticos: se ha producido un cambio espectacular en los últimos años. La inmensa mayoría de los pacientes con una DI padecen un trastorno genético, habitualmente estable, no progresivo. Los estudios genéticos básicos han pasado de ser el cariotipo, que generalmente revela escasa información, a la hibridación genómica comparada (CGH-arrays) y a una secuenciación exómica-genómica si es necesario u otras pruebas genéticas de otro tipo.
- Otros exámenes: el EEG es una prueba en determinadas situaciones de sospecha según determine el médico. Otros estudios deberán realizarse dependiendo del caso.
0 comentarios - 202017nov
Retraso psicomotor en la infancia (III Parte)

Evaluación del desarrollo psicomotor (DPM)
"El Pediatra juega un papel trascendental en el diagnóstico precoz del retraso psicomotor".
Los controles periódicos de salud en los primeros años de vida, van a proporcionar al Pediatra un momento extraordinario para valorar el DPM del niño en cada momento, así como la evolución cognitiva, social, motora, entre otras esferas, que presentará en los primeros años de vida.
Los programas de seguimiento del niño sano permiten la evaluación transversal y evolutiva del niño. Para facilitar este seguimiento, el Pediatra puede hacer uso de diferentes escalas de desarrollo. Ninguna de las escalas de desarrollo tiene un correlato fiable con el cociente intelectual del niño mayor. Algunas de las que se usan son la Escalas de Desarrollo Infantil de Bayley –BSID-, que evalúa el desarrollo infantil desde el nacimiento hasta los 2,5 años o el Test de Screening de Desarrollo de Denver –DDST-. Posiblemente la escala más empleada. Se trata más de un registro o cuestionario que una escala de desarrollo. Valora cuatro áreas: motor-gruesa, motor-fina, personal-social y lenguaje. En sus diferentes versiones, registra el desarrollo en estas áreas hasta los 4 años de edad y el Test de Haizea-Llevant. Similar al DDST en su sistema de evaluación y estimación de áreas comprometidas. Elaborada de forma específica en niños españoles hasta los 4 años. La TABLA I muestra hallazgos que, típicamente suelen ser normales, durante la evolución de los niños.
Diagnóstico etiológico
"La historia clínica y la exploración física son los apartados más importantes en la evaluación etiológica del retraso psicomotor (RPM)".
Anamnesis
La historia clínica debe ser completa. Se debe recoger de forma detallada el desarrollo psicomotor del paciente, no sólo el desarrollo motor. En el caso de un estancamiento o involución, deben anotarse, entre otras, la edad de comienzo, las áreas afectadas, los síntomas acompañantes si existieron y las causas atribuidas por los padres u otros profesionales.
Dentro de este apartado se reflejarán igualmente los antecedentes personales, control del embarazo, infecciones, características del parto, edad gestacional, instrumentación, etcétera.
La recogida de datos relacionados con el periodo neonatal aporta de nuevo una información trascendental (Apgar, peso al nacimiento, cuidados neonatales…), el resultado de screening metabólico, la presencia de hipotonía o crisis en los primeros días de vida, los problemas respiratorios, y otros problemas.
En relación a los antecedentes personales posteriores no se obviarán aquellos trastornos o enfermedades que puedan tener relación con la situación a estudio: crisis epilépticas (con/sin fiebre), meningoencefalitis, traumatismos craneoencefálicos graves, cardiopatías, entre muchas otras. Finalmente se añadirán los antecedentes familiares. Debemos intentar obtener un árbol genealógico amplio, pero más centrado en padres, abuelos y hermanos, en el que se haga constar los posibles antecedentes llamativos.
Exploración física
Debe iniciarse por un examen general que incluya entre otros la exploración de rasgos dismórficos (TABLA II), aunque sean menores, el perímetro craneal (fundamental), el desarrollo ponderoestatural, las características cutáneas, el desarrollo óseo, la presencia de visceromegalias, y cualquier otro dato que nos llame la atención.
El Pediatra, y especialmente el neurólogo infantil, no deben temer la descripción de rasgos que le resultan inicialmente anormales. Igualmente, no debe obviarse la obtención de imágenes-fotografías del niño o familia, ante la presencia de rasgos pecuilares, o por otros motivos. En ocasiones una descripción fenotípica detallada es la que orienta el diagnóstico. En otras ocasiones el desarrollo ponderoestatural apoya un diagnóstico de sospecha; la anotación de la talla-peso-perímetro craneal desde edades precoces puede orientar al diagnóstico. La identificación de anomalías menores y mayores resulta trascendental en estos casos. Dentro del examen por sistemas, algunas alteraciones podrán sugerir la etiología de base. La presencia de trastornos pigmentarios cutáneos puede apuntar hacia trastornos neurocutáneos frecuentes como la neurofibromatosis (fig. 1), la hipomelanosis de Ito o la esclerosis tuberosa, u otros menos frecuentes como la enfermedad de von Hippel-Lindau o la incontinentia pigmenti (fig. 2); la fotosensibilidad podrá orientar hacia la enfermedad de Hartnup, el exantema malar hacia la homocistinuria...entre otras muchas posibilidades. Las alteraciones del cabello pueden ser relevantes (por ejemplo, en la enfermedad de Menkes, en el hipotiroidismo, etcétera). La presencia de hepatoesplenomegalia apuntará hacia mucopolisacaridosis, esfingolipidosis, glucogenosis, entre otras (TABLA III).
Tras abordar un examen físico completo, se debe proceder a la exploración neurológica igualmente completa, valorando cualquier signo focal presente, asimetrías en el examen (Fig. 3), no obviando el examen craneal, la impresión subjetiva del nivel cognitivo, entre otras variables. De igual modo, Se debe hacer un fondo de ojo, un examen auditivo y visual si se precisara. La audiometría convencional, la discriminación visual o auditiva, la campimetría por confrontación son medidas realizables en cualquier consulta pediátrica, si bien complejas en el niño de corta edad.
La historia clínica y la exploración física completa y minuciosa, serán las que deberán orientar al diagnóstico, y a la consecuente realización de las exploraciones complementarias oportunas. En éstas profundizaremos en la próxima revisión del blog.
Figura 1. Manchas color "café con leche" características de la neurofibromatosis tipo 1.
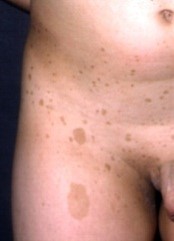
Figura 2. Lesiones características de la incontinentia pigmenti.
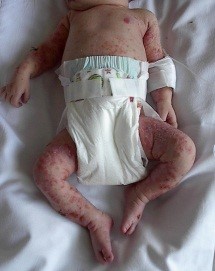
Figura 3. Marcha de inicio asimétrica de niño con hemiparesia izquierda.

TABLA I: VARIACIONES DE LA NORMALIDAD SIN CARÁCTER PATOLÓGICO
Pinza entre dedo pulgar y medio
Marcha de pie sin pasar por la fase de gateo
Desplazamiento sentado sobre nalgas o apoyando una rodilla y el pie de la otra extremidad o rodar sobre sí mismo
Marcha de puntillas primeras semanas o meses tras el inicio de la deambulación
Rotación persistente de la cabeza.
Retraso simple de la marcha con signo de ‘sentarse en el aire’
Tartamudeo fisiológico: entre los 2-4 años
Dislalias fisiológicas: hasta los 4-5 años
Otras: para neuropediatría
TABLA II. Malformaciones menores y mayores según localización (ejemplos)
Menores
Mayores
Cutáneas
Nevus
Hemangiomas
Manchas café con leche
Alopecia congénita
Hipertricosis
Craneales
Occipucio plano
Frente promiente
Craneosinostosis
Fístulas branquiales
Faciales
Hipertelorismo
Orificios nasales antevertidos
Boca en carpa
Orejas de implantación baja
Anoftalmia
Labio leporino
Atresia meato auditivo
Torácica
Tórax en tonel
Mamilas separadas
Malformaciones cardiovasculares
Abdominales
Hernia umbilical
Diastasis de rectos
Distensión abdominal
Malrotación o atresia intestinal
Onfalocele
Urogenital
Mínimo hipospadias
Teste en ascensor
Genitales ambigüos
Criptorquidia
Epispadias
Esqueléticas
Cubitus valgo
Genu recurvatum
Fosita sacra
Pie equinovaro
Hemivértebras
Polidactilia
SNC
Displasias corticales
Meningocele
TABLA III. Afectación de diferentes órganos o sistemas según trastorno metabólico
Afectación hepática y/o esplénica
Con hepatoesplenomegalia:
Esfingolipidosis, mucolipidosis, mucopolisacaridosis, trastornos peroxisomales, galactosemia…
Con ictericia y fallo hepático:
Enfermedad de Wilson, enfermedad de Niemann-Pick C, síndrome de Alpers, galactosemia…
Cardiopatía
Glucogenosis tipo 2, enfermedad de Friedreich, enfermedad de Refsum, enfermedad de Fabry, mucopolisacaridosis, homocistinuria…
Nefropatía
Síndrome de Lowe, enfermedad de Zellweger, enfermedad de Fabry, enfermedad de Lesch-Nyhan, galactosemia, acidemia isovalérica…
Afectación esquelética
Mucolipidosis, mucopolisacaridosis, sialidosis, enfermedad de Zellweger, enfermedad de Lowe, enfermedad de Refsum…
Afectación cutánea
Enfermedad de Hartnup, síndrome de Cockayne, xeroderma pigmentoso, fucosidosis, déficit de biotinidasa…
Anomalías hematológicas
Anemia:
Enfermedad de Gaucher, enfermedad de Fabry…
Trombocitopenia:
Acidemia isovalerica, acidemia propiónica, enfermedad de Wilson…
Afectación respiratoria
Enfermedad de Menkes, glucogenosis tipo II, enfermedad de Farber…
Afectación digestiva
Abetalipoproteinemia, MELAS, porfiria aguda intermitente…
- 202011nov
TDAH y COVID-19
Los pacientes con TDAH son más vulnerables a la infección por COVID-19 y el tratamiento farmacológico disminuye la probabilidad de infección de pacientes con TDAH

La enfermedad por coronavirus denominada COVID-19 es una pandemia que se ha expandido por todo el planeta y a todos los países procedentes de Wuham (China) desde finales del año 2019.
Las medidas efectivas para impedir el contagio incluyen evitar el contacto próximo con otras personas, manteniendo al menos una distancia de 2 m. Portar una mascarilla en público y en lavado de manos frecuente son otras medidas importantes. La gente debe permanecer en casa y aislarse cuando se padecen síntomas leves tales como cefalea, fiebre o incluso una tos leve.
Los criterios diagnósticos del trastorno por déficit de atención / hiperactividad (TDAH) incluyen síntomas que tienden a incrementar el riesgo de la transmisión del virus. Los criterios de la tensión incluyen problemas para prestar atención a los detalles o cometer errores por despiste, no estar atentos cuando otras personas les hablan directamente, y dificultades en la concentración y en apuntar en la agenda instrucciones vitales para el aprendizaje, o traer a casa los materiales necesarios para realizar adecuadamente las tareas.
Los criterios de hiperactividad e impulsividad incluyen la inquietud, el movimiento de manos y pies frecuente, especialmente durante la sedestación, y de forma muy específica levantarse de la silla o el asiento en situaciones en las que deberían permanecer sentados, especialmente en clase.
Eugene Merzon, Iris Manor y colaboradores han realizado un estudio que incluyó un análisis multivariante de las variables estudiadas, con más de 14.000 pacientes registrados en los servicios de salud denominados Leumit (Israel) entre febrero y abril de este año. Un total de 1.416 pacientes fueron COVID +. Estos pacientes fueron significativamente más jóvenes, con más frecuencia eran varones y tenían Ratios de TDAH más altos (16% versus 11%) que los pacientes que eran COVID negativo. Cuando no estaban siendo tratados farmacológicamente (93% mediante psicoestimulantes como el metifenidato o la lisdexanfetamina) de sus síntomas TDAH, el porcentaje de pacientes COVID positivo era de 75 % (13% del total de pacientes COVID +). Sin embargo, los pacientes que estaban siendo tratados adecuadamente para mitigar los síntomas del TDAH (al menos tres prescripciones consecutivas durante un año) no tenían mayor riesgo que los pacientes controles para padecer COVID-19 (por el contrario, tenían menos riesgo de padecer muchas otros trastornos, tales como depresión, ansiedad, demencia, hipertensión, entre otros, estos últimos, probablemente, porque al saberse más sensibles a la infección se quedaban más en casa), bajando el porcentaje a un 24% (3% del total de pacientes COVID+).
Por lo tanto, el ratio de pacientes con TDAH COVID-19 positivos tratados versus no tratados era de uno a tres.
El estudio, con un tamaño de la muestra importante y una metodología bien desarrollada, parece mostrar de una forma muy clara y convincente que los pacientes no tratados con TDAH tienen mayor riesgo de padecer la infección por COVID-19 y que los pacientes tratados adecuadamente con fármacos para el TDAH aminoran este riesgo.
Es posible que esta asociación pueda deberse a los síntomas centrales del trastorno y secundariamente a riesgos relacionados con comportamientos tales como atender a fiestas masivas, reunirse en pandillas, o no cumplir con las distancias de seguridad recomendadas por el gobierno de Israel. De hecho, el TDAH (no tratado farmacológicamente) fue el único trastorno codificado en el manual DSM-5 que se identificó como un factor de riesgo significativo para aumentar la probabilidad de infección por COVID-19.
Por lo tanto, los pacientes con TDAH son más vulnerables a la infección por COVID-19 y el tratamiento farmacológico disminuye la probabilidad de infección en los pacientes con TDAH.
Bibliografía: https://journals.sagepub.com/doi/10.1177/1087054720943271
- 20203nov
Retraso psicomotor en la infancia (II Parte)

Clasificación etiológica
"Un retraso psicomotor no siempre es patológico o anormal, pero puede ser también la antesala de graves problemas del desarrollo físico y cognitivo del niño"
1. Variante de la normalidad.
Los márgenes de la normalidad para numerosos hitos son amplios. En ocasiones, especialmente en RPM parciales, encontramos pacientes completamente sanos, que se "escapan" de los márgenes señalados como "normales" para la población a estudio, los signos más frecuentes aparecen en la TABLA III. Por ejemplo, un tercio de los niños no gatea nunca, por lo que es un signo más "tranquilizador" en su presencia que de "alarma" en su ausencia.
Dos circunstancias especiales en este sentido son el recién nacido prematuro (RNPT) y el niño ingresado-encamado. El RNPT alcanzará los hitos lógicos del DPM más tarde que los demás; para valorar la normalidad del desarrollo en estos niños, deberá emplearse la edad corregida, es decir la edad que el niño tendría si hubiera nacido en la fecha prevista del parto (edad corregida= "edad cronológica medida en semanas o meses"-"número de semanas o meses de prematuridad"). Esta corrección es especialmente necesaria en los primeros 24 meses. Por otro lado, la prematuridad es un factor de riesgo para los problemas del desarrollo y el aprendizaje, por lo que el DPM deberá ser vigilado estrechamente. El niño ingresado o encamado durante tiempos largos durante el 1º-2º año de vida, puede igualmente mostrar un leve retraso o estancamiento del desarrollo motor. En estos niños se puede sumar el RPM a déficit asociados en el desarrollo por la patología que justificó el ingreso hospitalario.
2. Hipoestimulación.
Los niños pobremente estimulados o institucionalizados pueden mostrar un claro RPM en los primeros meses de la vida. Esta circunstancia es generalmente normalizable. Sin embargo, cuando la hipoestimulación es severa y mantenida, como sucede en niños adoptados del este de Europa, puede justificar, junto a otros factores de riesgo, futuros problemas del neurodesarrollo.
3. Déficit neurosensorial
Los problemas sensoriales, especialmente auditivos o visuales, pueden ser causa de un RPM. Es habitual que la patología auditiva severa se asocie con retrasos del lenguaje, la comunicación, e incluso con conductas de aislamiento que pueden recordar trastornos generalizados del desarrollo. Por ello deben descartarse en todos los casos, bien por audiometría, bien por potenciales evocados auditivos del tronco encefálico (PEATE) si el niño no colabora o es muy pequeño. Los niños con trastornos auditivos pueden mostrar un desarrollo del lenguaje normal los primeros 6 meses de vida (ruidos, risas, balbuceos…) con interrupción del mismo por ausencia de feedback ambiental. Es excepcional que la hipoacusia leve uni o bilateral justifique un verdadero retraso del lenguaje; del mismo modo, no debe justificarse el retraso del lenguaje a otitis recurrentes.
Los problemas visuales pueden igualmente asociarse a problemas de la coordinación, manipulación… Estos generalmente están relacionados con el componente visual-sensorial, no con el motor; un estrabismo o nistagmus puede ser un signo de un trastorno neurológico de base, pero no la causa de un RPM
4. Anticipación de un trastorno específico del desarrollo
Los trastornos del desarrollo de la coordinación y los trastornos de la comunicación, tienden a anticiparse por RPM con afectación predominantemente motora y del lenguaje respectivamente.
5. Anticipación de un trastorno motor
La parálisis cerebral infantil (PCI) tiende a manifestarse en los primeros 18-24 meses de vida por un RPM global o predominantemente motor. Aunque PCI se define como un trastorno motor, crónico, de comienzo precoz y no progresivo, las manifestaciones clínicas pueden ser cambiantes y más invalidantes durante el desarrollo del niño. Puede además acompañarse de problemas sensoriales (visuales hasta en el 50% y auditivos hasta en el 15% de los casos), epilepsia (25-35%) que puede condicionar el propio desarrollo global o discapacidad intelectual (DI) -retraso mental- (hasta en el 50% de los niños).
En este apartado debemos incluir las miopatías, tanto las congénitas como las distrofias musculares, que pueden manifestarse con carácter estático o progresivo respectivamente, y a veces acompañadas de retraso cognitivo. Igualmente, no deben obviarse otras enfermedades como la atrofia muscular espinal que se manifestará en los primeros meses de vida, o algunas neuropatías genéticas, que podrán hacerlo en los 3-4 primeros años, en forma de retraso motor, hipotonía o torpeza.
6. Anticipación de una discapacidad intelectual –DI- (retraso mental)
Generalmente la mayoría de los pacientes con DI (este término ha ido sustituyendo al de retraso mental), han tenido al menos cierto RPM. En ocasiones, las DI leves se anticipan por leves RPM o RPM parciales, que pasan desapercibidos para la familia o el médico.
Es un trastorno plurietiológico; habitualmente de causa genética. Estos últimos años el diagnóstico etiológico ronda el 50% de los casos leves y el 80% de los graves gracias a los espectaculares avances en el diagnóstico genético estos últimos 3-4 años.
Es frecuente que los pacientes con DI asocien otros problemas neurológicos que contribuyen de forma desfavorable en el DPM. Algunos estudios refieren la asociación a encefalopatías motoras en el 7% de los pacientes, epilepsia en el 10%, alteraciones neurosensoriales en el 7% o autismo en el 2-3%. Estas asociaciones se muestran más intensas cuanto menor es el CI.
7. Anticipación de un trastorno del espectro autista (TEA)
Caracterizado eminentemente por una alteración de la socialización, la comunicación y un patrón de intereses restringidos y comportamientos estereotipados, se puede manifestar con un desarrollo lento o atípico. Estos problemas pueden acompañarse de cierta torpeza o hipotonía en los primeros meses de vida (a menudo debido a la causa genética subyacente del trastorno), pero suele expresarse en los primeros meses/años de vida con una alteración cualitativa y/o cuantitativa del lenguaje y ser indiferenciable inicialmente de un trastorno específico del lenguaje (TEL) con afectación de la comprensión del mismo. En estos casos lo más importante es derivar al neuropediatra e instaurar una intervención precoz adecuada mientras se realizan las pruebas complementarias oportunas. La ausencia de un diagnóstico específico no puede demorar la derivación de un niño con sospecha de TEA/TEL a un centro de atención temprana especialista en trastornos de la comunicación. Separar este grupo de los anteriores se muestra complejo, dada de nuevo su comorbilidad con otros trastornos o enfermedades. Hasta el 90% de los casos de TEA grave pueden tener DI, y epilepsia hasta en la mitad de los casos (especialmente si tienen DI), alteraciones visuales o auditivas leves que pueden condicionar el DPM, etcétera.
Tabla. Prevalencia de las principales causas del RPM global o parcial.
Sordera
0,1%
Ceguera
1,5-6/10000
Trastorno del desarrollo de la coordinación
6%
Trastorno de la comunicación
4-6%
Parálisis cerebral infantil
0,2%
Retraso mental (discapacidad intelectual)
1%
Autismo
1%
Sobre este blog
Blog sobre los temas relacionados con la neuropedciatría: déficit de atención, hiperactividad, epilepsia, cefaleas, tics, encefalitis, problemas escolares, etc.

Archivo del blog
 2.024
2.024
- 2.023
- 2.022
- 2.021
- 2.020
- 2.019
- 2.018
- Diciembre
- Noviembre
- Octubre
- Julio
- Mayo
- Abril
- Febrero
- Recomendaciones generales y específicas para el trastorno por déficit de atención/hiperactividad -TDAH- (III). Recomendaciones específicas.
- El Dr. Daniel Martín Fernández-Mayoralas ganador del “Concurso de Casos Clínicos sobre el abordaje farmacológico de pacientes con TDAH" organizado por el Grupo Saned
- Enero
- 2.017
- 2.016
Últimas entradas
- Síndrome cognitivo afectivo del cerebelo: Un diagnóstico a tener en cuenta.
- Síndrome de Pitt-Hopkins
- Tratamiento Cognitivo Conductual en adolescentes con Trastorno por Déficit de Atención con/sin Hiperactividad (TDAH)
- Ejercicio y TDAH
- Empoderando a los Docentes: Estrategias para detectar y abordar dificultades en el aula. (III)
Colaboraciones




La finalidad de este blog es proporcionar información de salud que, en ningún caso sustituye la consulta con su médico. Este blog está sujeto a moderación, de manera que se excluyen de él los comentarios ofensivos, publicitarios, o que no se consideren oportunos en relación con el tema que trata cada uno de los artículos.
Quirónsalud no se hace responsable de los contenidos, opiniones e imágenes que aparezcan en los "blogs". En cualquier caso, si Quirónsalud es informado de que existe cualquier contenido inapropiado o ilícito, procederá a su eliminación de forma inmediata.
Los textos, artículos y contenidos de este BLOG están sujetos y protegidos por derechos de propiedad intelectual e industrial, disponiendo Quirónsalud de los permisos necesarios para la utilización de las imágenes, fotografías, textos, diseños, animaciones y demás contenido o elementos del blog. El acceso y utilización de este Blog no confiere al Visitante ningún tipo de licencia o derecho de uso o explotación alguno, por lo que el uso, reproducción, distribución, comunicación pública, transformación o cualquier otra actividad similar o análoga, queda totalmente prohibida salvo que medie expresa autorización por escrito de Quirónsalud.
Quirónsalud se reserva la facultad de retirar o suspender temporal o definitivamente, en cualquier momento y sin necesidad de aviso previo, el acceso al Blog y/o a los contenidos del mismo a aquellos Visitantes, internautas o usuarios de internet que incumplan lo establecido en el presente Aviso, todo ello sin perjuicio del ejercicio de las acciones contra los mismos que procedan conforme a la Ley y al Derecho.




© 2024 Quirónsalud - Todos los derechos reservados
