Quirónsalud







Blog del Dr. Daniel Martín Fernández-Mayoralas. Neurología. Complejo Hospitalario Ruber Juan Bravo y Hospital Universitario Quirónsalud Madrid
- 202328abr
Redes sociales y salud mental
Con la colaboración de María del Rosario Campos Díaz. Psicóloga Clínica Infanto- Juvenil y responsable del Centro Cogniciona (Pozuelo de Alarcón)
Las redes sociales son aplicaciones web cuya finalidad es la comunicación entre individuos, la transmisión de información, y sabemos que la información es poder. Sin embargo, a pesar de las ventajas de su uso, por ejemplo, a nivel laboral, y teniendo en cuenta que conectarse a ellas forma parte de la conducta normal y cotidiana de la vida moderna (Andreassen, 2015), diversos estudios empíricos llegan a la conclusión de que las personas que emplean menos tiempo en redes sociales tienen mejor salud mental (Moreno y otros, 2011).

Dichas investigaciones evidencian una correlación directa entre el uso de redes sociales y la ansiedad, al igual que con la depresión. Además, está demostrado que a mayor cantidad de tiempo de uso de redes sociales, mayor probabilidad de padecer depresión y ansiedad.
El impacto en nuestro estado emocional y, más aún, en el de nuestros niños/as y adolescentes va aún más allá. Así, en el trabajo de investigación "Tiempos Modernos: Redes Sociales y Ansiedad", de Laura Vanessa García Gualdrón (asesorada por Mario Andrés Ernesto Martín Padilla), en 2015, se encontró que el uso de las redes sociales en población juvenil se encuentra caracterizada por una fuerte dependencia psicológica, e impulsividad, lo que repercute en el área social y personal de los usuarios, causando también un mal uso del tiempo.
Mucho se ha escrito ya sobre la adicción a las redes sociales, que se relaciona de forma negativa con el nivel de autoestima, y de forma significativa con el nivel de ansiedad, de manera que los estudiantes con mayor adicción a dichas redes manifiestan también un mayor nivel de ansiedad, como se constata en el estudio publicado en el siguiente artículo: Portillo-Reyes, V. Ávila-Amaya, J. A., Capps, J. W. (2021). Relación del Uso de Redes Sociales con la Autoestima y la Ansiedad en Estudiantes Universitarios. Enseñanza e Investigación en Psicología, 3(1), 139-149.
Otras consecuencias de su mal uso serían: amistades débiles y superficiales, exclusión, estrés, depresión, adicción y alteraciones en los patrones de sueño, envidia, vinculaciones irreales, entre otras (Drahošová y Balco, 2017). El uso excesivo de redes sociales también puede generar alteraciones emocionales por la visualización de información de alto impacto o de estándares y prototipos de belleza casi imposibles.
En relación a ello, señalar el artículo publicado en la revista Behavioral Psychology / Psicología Conductual, Vol. 30, Nº 3, 2022, pp. 677-691 https://doi.org/10.51668/bp.8322305s
 : EFECTO DE LA EXPOSICIÓN A IDEALES DE DELGADEZ EN LAS REDES SOCIALES SOBRE LA AUTOESTIMA Y LA ANSIEDAD, de Blanca Rodríguez-Suárez1 , José Manuel Caperos1 y José Ángel Martínez-Huertas1,2 1 UNINPSI, Universidad Pontificia Comillas; 2 Universidad Nacional de Educación a Distancia (España), que resume que el uso de redes sociales está relacionado con la aparición de trastornos de la conducta alimentaria (TCA). Los autores encuentran una diminución de la autoestima en el grupo expuesto a imágenes de carga comparativa alta y un aumento de la ansiedad. Concluyen que efecto de las imágenes sobre la autoestima está completamente mediado por el incremento en la ansiedad.
: EFECTO DE LA EXPOSICIÓN A IDEALES DE DELGADEZ EN LAS REDES SOCIALES SOBRE LA AUTOESTIMA Y LA ANSIEDAD, de Blanca Rodríguez-Suárez1 , José Manuel Caperos1 y José Ángel Martínez-Huertas1,2 1 UNINPSI, Universidad Pontificia Comillas; 2 Universidad Nacional de Educación a Distancia (España), que resume que el uso de redes sociales está relacionado con la aparición de trastornos de la conducta alimentaria (TCA). Los autores encuentran una diminución de la autoestima en el grupo expuesto a imágenes de carga comparativa alta y un aumento de la ansiedad. Concluyen que efecto de las imágenes sobre la autoestima está completamente mediado por el incremento en la ansiedad.En la actualidad, las redes sociales cubren algunas de nuestras necesidades en el mundo de la apariencia, como las del reconocimiento, la necesidad de afecto (muchas veces, medida en función de los likes, los retwit, o el número de visualizaciones de vídeos colgados). Las redes te dan la posibilidad de mostrar una imagen de ti que no puedes ser en la vida real e, incluso, el desarrollar distintas identidades, con el peligro que ello supone en una personalidad que se está forjando.
Por otra parte, "las redes no duermen", por lo que algunos jóvenes sufren angustia por no poder responder de inmediato a mensajes durante la noche o durante su asistencia a clases u otras obligaciones, o bien, sí los contestan, a costa de su higiene del sueño y de la repercusión negativa en sus estudios. Por no hablar de lo problemas de conducta en algunos cuando sus padres tratan de poner límites en este sentido.
En 2018, Marina Díaz- Marsá, Presidenta de la Sociedad de Psiquiatría de Madrid, ya declaraba que cada vez hay más diagnósticos en jóvenes depresivos que pasan horas y horas en redes sociales.
En consulta, cada vez más a menudo, además de lo comentado, los profesionales de la salud mental vemos jóvenes víctimas de ciberacoso o de pederastas que contactan con ellos a través de las redes. Los más vulnerables son los/las menores que ya presentan un trastorno de base, como un TDAH o un TAS, de manera que podemos casi hablar de patología dual.
Todo ello hace imprescindible la prevención, por lo que a continuación se explicitan algunas pautas concretas para un buen uso de la redes, principalmente en los más jóvenes:
- Controlar el tiempo de conexión a Internet.
- No creer por completo todo lo que se publica en estos medios.
- Identificar grupos sociales con gustos o intereses similares a los propios.
- No dedicar demasiado tiempo a visualizar publicaciones de otros.
- Evitar pensar de forma irracional, por ejemplo, que es necesario hacer publicaciones que llamen la atención para ser aceptado/a.
- Participar en actividades sociales fuera de la red.
- Realizar actividad física con frecuencia.
- Fomentar otras aficiones y actividades, tanto individuales como en equipo (pero de manera presencial).
- Trabajar la propia autoestima, independiente de la opinión de los demás.
Para concluir, como no podía ser de otra forma, se recomiendas los siguientes enlaces web de cara a la seguridad en Internet:
— Estrategia Nacional de Ciberseguridad 2019. https://www.dsn.gob.es/es/documento/estrategia-nacional-ciberseguridad-2019
.Redes sopciales - RR.SS. - salud mental - ansiedad - depresión - dependencia psicológica - impulsividad - autoestima - higiene del sueño - TDAH - TAS - Quirónsalud - Hospital Universitario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Daniel Martín Fernández Mayoralas - neuropediatra1 comentario - 202227abr
El síndrome de kabuki (KS)
El síndrome de Kabuki (KS), es una patología con múltiples anomalías. Las más frecuentes: características faciales peculiares que remedan el maquillaje Kabuki -fisuras palpebrales largas y eversión de los párpados, que consiste en que éstos se ven como si se les hubiera dado la vuelta y se observa la parte interior, es decir, la parte que está en contacto con el ojo-, anomalías esqueléticas, engrosamiento de las yemas de los dedos y talla baja, asociadas a discapacidad intelectual. Pese a que se pensaba que era un síndrome muy raro, actualmente sabemos que afecta a cerca de una cada 30.000 personas aproximadamente, por lo que es una causa relativamente común de discapacidad intelectual, a tener siempre en cuenta, de ahí el interés del presente post.
El gen KMT2D, también conocido como MLL2, proporciona instrucciones para producir una enzima llamada metiltransferasa 2D específica de la lisina, que se encuentra en muchos órganos y tejidos del cuerpo. Esta enzima funciona modificando las histonas, agregándolas un grupo metilo (metilación), de este modo, las histonas metiltransferasas controlan la actividad de ciertos genes a nivel de empaquetamiento de la cromatina (la forma en la que se presenta el ADN en el núcleo celular). Así, la enzima codificada por KMT2D parece activar ciertos genes que son importantes para el desarrollo. Se han identificado diversas y múltiples mutaciones en el gen KMT2D, en personas con KS. El KDM6A, ligado al cromosoma X, puede ser responsable con menor frecuencia del KS, siendo más grave en varones. Existe la posibilidad de realizar el diagnóstico genético de esté síndrome en la práctica clínica, a través de una técnica denominada exoma.
Las anomalías estructurales en el KS pueden incluir lo siguiente:
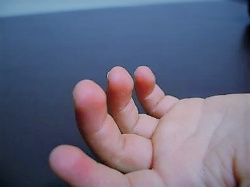
- Yemas de los dedos fetales persistentes (engrosamiento de las yemas de los dedos de las manos); se consideran una de las cinco manifestaciones cardinales del KS y, por lo tanto, se encuentran en una gran proporción de las personas afectadas.
- Oftalmológicas, incluyendo ptosis y estrabismo. Curiosamente, como resultado de la eversión del párpado inferior, los niños con KS pueden mostrar un lagrimeo excesivo, que generalmente no es un problema importante.
- Otológicas (una pista de diagnóstico potencialmente útil es que la mayoría de las personas con KS tienen orejas prominentes y en forma de copa. Los hoyos en el trago y en la región posterior de los pabellones auriculares también son relativamente comunes). La sordera neurosensorial es rara, aunque las otitis medias son comunes y a veces pueden producir pérdida de audición.
- Bucodentales: Labio y/o paladar hendido (un tercio de los niños). Anomalías dentales que incluyen dientes muy separados e hipodoncia (lo que significa que puede haber a veces incisivos superiores laterales ausentes, o incisivos inferiores ausentes, o molares superiores ectópicos y/o segundos premolares faltantes).
- Defectos cardíacos congénitos (cerca de la mitad de los casos, el más frecuente la coartación de aorta).
- Gastrointestinales, incluida la atresia anal. Las más frecuentes están relacionadas con hipotonía, mala coordinación oromotora y dificultades para tragar.
- Genitourinarias, incluyendo criptorquidia (testículo no descendido) en varones.
- Endocrinológicas, incluyendo telarquia prematura (aparición del botón mamario por primera vez en la mujer). En la adolescencia y la edad adulta, más de la mitad de las personas con KS desarrollan obesidad. La deficiencia del crecimiento posnatal es evidente a los 12 meses de edad. La falta de un crecimiento acelerado típico durante la pubertad exacerba la baja estatura. Pueden responder a tratamiento con hormona de crecimiento (GH). Otros hallazgos: hiperinsulinismo, insuficiencia suprarrenal, deficiencia combinada de hormona pituitaria, diabetes insípida, deficiencia franca de hormona de crecimiento, hipotiroidismo, disfunción ovárica primaria, verdadera pubertad precoz.
- Aumento susceptibilidad a enfermedades autoinmunes. No hay evidencia de aumento de cáncer.
- Neurológicas:
- Hipotonía, aunque la hiperlaxitud articular es la regla, lo que afecta al tono pasivo.
- Luxaciones o subluxaciones articulares, que afectan especialmente a las caderas, las rótulas y los hombros. No son infrecuentes, pero como en la mayoría de las condiciones con laxitud articular, este hallazgo mejora con la edad.
- Epilepsia o crisis epilépticas. No suelen ser de difícil control.
- Discapacidad intelectual, generalmente en el rango leve a moderado, en la mayoría de las personas; sin embargo, se han publicado informes de individuos con variantes patogénicas en KMT2D o KDM6A que tienen niveles de coeficiente intelectual superiores a 70. La mayoría de las personas con KS pueden hablar y caminar adecuadamente. No se ha identificado ningún perfil lingüístico específico. Sin embargo, todos los subdominios del lenguaje, incluidos la sintaxis, la morfología, la pragmática y la semántica, pueden verse afectados. En las pruebas neuropsiquiátricas formales, las personas con KS tienden a obtener mejores puntuaciones en las áreas de comprensión de vocabulario y memoria de trabajo y más bajas en las áreas de razonamiento no verbal y velocidad de procesamiento. Las personas con KS tienden a ser descritas como agradables y extrovertidas. En un subconjunto de individuos afectados existe TDAH. Rara vez se han informado otros trastornos: ansiedad, trastornos del espectro autista, trastornos de conducta, trastornos del sueño, etcétera.
El médico especialista, la mayor parte de las veces el Neuropediatra, debe decidir las pruebas a realizar en cada caso (ecografía renal/abdominal, EEG, interconsulta a cardiología, etc.) según los síntomas y signos detectados y/o esperados en cada paciente, de forma individualizada.
BIBLIOGRAFÍA:
Adam MP et al. Kabuki Syndrome. 2011 Sep 1 [Updated 2021 Jul 15]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022.
Banka S et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. European Journal of Human Genetics (2012) 20, 381–388.
Lepri FR. Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature. Int. J. Mol. Sci. 2018, 19, 82; doi:10.3390/ijms19010082.
- 202128sep
El desarrollo del lenguaje: lo esperable en el desarrollo normal
Colaboración del Dr. Daniel Martín y de Ana Alás Rupérez
Ana Alás Rupérez es graduada en Logopedia y dedica su labor profesional a la evaluación, diagnóstico e intervención de alteraciones del lenguaje y/o la comunicación. Coordina la Unidad de Logopedia del Hospital Universitario Quirónsalud Madrid y del Hospital Quirónsalud San José, además de dirigir un centro privado (www.centrocomunica.com
), dedicado al abordaje de trastornos del neurodesarrollo. 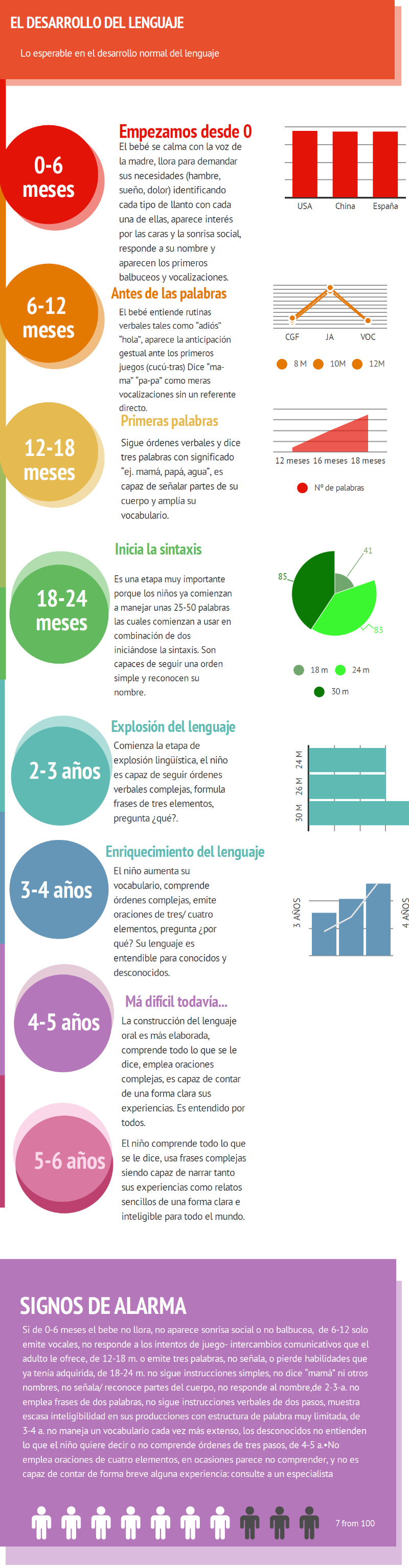 Desarrollo del lenguaje - desarrollo de la comunicación - lenguaje - comunicación - logopedia - alteraciones del lenguaje - alteraciones de la comunicación - Quirónsalud - complejo hospitalario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Daniel Martín Fernández-Mayoralas - Ana Alás Rupérez - neuropediatra0 comentarios
Desarrollo del lenguaje - desarrollo de la comunicación - lenguaje - comunicación - logopedia - alteraciones del lenguaje - alteraciones de la comunicación - Quirónsalud - complejo hospitalario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Daniel Martín Fernández-Mayoralas - Ana Alás Rupérez - neuropediatra0 comentarios - 20217abr
Tratamiento del insomnio en niños con trastornos del espectro autista (TEA). Hincapié en la melatonina.
El TEA es un trastorno del neurodesarrollo que afecta a algo más de uno de cada 100 niños, especialmente varones. Dado que engloba más de 3.000 causas (etiologías) diferentes, la mayor parte de ellas relacionadas con mutaciones genéticas, parece apropiado definir el TEA como un síndrome heterogéneo multicausal. El Manual diagnóstico y estadístico de los trastornos mentales, quinta edición (DSM-5), ha modificado los criterios exigidos para la clasificación comparado con clasificaciones previas, reduciendo éstos a dos dimensiones de síntomas: comunicación social (afectación de la reciprocidad social y de la comunicación) e intereses restringidos y comportamientos repetitivos y restrictivos.

La resistencia a la hora de acostarse es un fenómeno conductual que se manifiesta mediante el rechazo a irse a la cama, posponerlo o requerir la presencia de uno de los padres al inicio del sueño. A pesar de tratarse de un tema poco tratado en ensayos clínicos aleatorizados controlados con placebo existe un buen estudio de clase II con niños con TEA e insomnio al inicio del sueño o insomnio de mantenimiento1. En este estudio se comprobó que el tratamiento con melatonina fue efectivo para reducir los síntomas del insomnio, mientras que la terapia cognitivo-conductual tuvo un impacto levemente positivo principalmente en la latencia del sueño, lo que sugiere que algunos aspectos conductuales podrían desempeñar un papel en la determinación del insomnio inicial1. Del mismo modo, otro estudio que valoró la latencia de inicio del sueño, esto es, la cantidad de tiempo desde que se apaga la luz hasta el inicio de la etapa de sueño dio como resultado una reducción media en la latencia de -33.1 minutos (95% IC, −43.5 a −22.6; I2 = 0%) para niños con TEA y alteración del sueño tratados con melatonina2. Otros estudios basados en colchones especiales no ofrecieron resultados de beneficio2.
En la práctica, se encuentran concentraciones variables de melatonina en las preparaciones de venta libre (internet, por ejemplo), de modo que la melatonina obtenida por prescripción en farmacias es más segura. Cuando se usa como hipnótico, la melatonina se administra generalmente 30 minutos antes de la hora de dormir. Debido a que la melatonina de liberación inmediata tiene una vida media corta (40 minutos), se asume que las formulaciones de liberación inmediata son más útiles para el insomnio de inicio del sueño y las formas de liberación controlada, para el mantenimiento del sueño.
Aunque no se conocen efectos secundarios graves, no se conoce del todo si hay efectos secundarios leves a muy largo plazo, por lo que la prescripción de la melatonina en el TEA debe sopesarse contra los daños del trastorno de sueño persistente para individuos con TEA y sus familias, que cuando son intensos, sobrepasan con mucho los potenciales efectos secundarios de la melatonina.
- 20213mar
Ortesis craneales
A menudo los pacientes nos preguntan sobre el mecanismo de funcionamiento de las ortesis craneales cuando existe plagiocefalia, braquicefalia o dolicocefalia.
Para las cuestiones básicos recomendamos acudir al primer capítulo sobre este tema, https://www.quironsalud.es/blogs/es/neuropediatra/plagiocefalia-tratamiento
.Los principios de la intervención mediante ortesis para la plagiocefalia en general se basan en proporcionar un contacto total en las áreas en las cuales se pretende frenar el crecimiento del cráneo para mejorar la deformidad. Se debe permitir un espacio entre la ortesis y el cráneo en las áreas donde se desea que la cabeza crezca. Existe una ventana de oportunidad crítica entre los tres y los nueve meses de edad, en algunos casos hasta los 12 meses de edad cuando la cabeza se está formando activamente. El casco, esto es la ortesis, siguiendo estas pautas va a conseguir que crecimiento cefálico se produzca con normalidad aliviando la deformidad.



Sobre este blog
Blog sobre los temas relacionados con la neuropedciatría: déficit de atención, hiperactividad, epilepsia, cefaleas, tics, encefalitis, problemas escolares, etc.

Archivo del blog
 2.024
2.024
- 2.023
- 2.022
- 2.021
- 2.020
- 2.019
- 2.018
- Diciembre
- Noviembre
- Octubre
- Julio
- Mayo
- Abril
- Febrero
- Recomendaciones generales y específicas para el trastorno por déficit de atención/hiperactividad -TDAH- (III). Recomendaciones específicas.
- El Dr. Daniel Martín Fernández-Mayoralas ganador del “Concurso de Casos Clínicos sobre el abordaje farmacológico de pacientes con TDAH" organizado por el Grupo Saned
- Enero
- 2.017
- 2.016
Últimas entradas
- Síndrome cognitivo afectivo del cerebelo: Un diagnóstico a tener en cuenta.
- Síndrome de Pitt-Hopkins
- Tratamiento Cognitivo Conductual en adolescentes con Trastorno por Déficit de Atención con/sin Hiperactividad (TDAH)
- Ejercicio y TDAH
- Empoderando a los Docentes: Estrategias para detectar y abordar dificultades en el aula. (III)
Colaboraciones




La finalidad de este blog es proporcionar información de salud que, en ningún caso sustituye la consulta con su médico. Este blog está sujeto a moderación, de manera que se excluyen de él los comentarios ofensivos, publicitarios, o que no se consideren oportunos en relación con el tema que trata cada uno de los artículos.
Quirónsalud no se hace responsable de los contenidos, opiniones e imágenes que aparezcan en los "blogs". En cualquier caso, si Quirónsalud es informado de que existe cualquier contenido inapropiado o ilícito, procederá a su eliminación de forma inmediata.
Los textos, artículos y contenidos de este BLOG están sujetos y protegidos por derechos de propiedad intelectual e industrial, disponiendo Quirónsalud de los permisos necesarios para la utilización de las imágenes, fotografías, textos, diseños, animaciones y demás contenido o elementos del blog. El acceso y utilización de este Blog no confiere al Visitante ningún tipo de licencia o derecho de uso o explotación alguno, por lo que el uso, reproducción, distribución, comunicación pública, transformación o cualquier otra actividad similar o análoga, queda totalmente prohibida salvo que medie expresa autorización por escrito de Quirónsalud.
Quirónsalud se reserva la facultad de retirar o suspender temporal o definitivamente, en cualquier momento y sin necesidad de aviso previo, el acceso al Blog y/o a los contenidos del mismo a aquellos Visitantes, internautas o usuarios de internet que incumplan lo establecido en el presente Aviso, todo ello sin perjuicio del ejercicio de las acciones contra los mismos que procedan conforme a la Ley y al Derecho.




© 2024 Quirónsalud - Todos los derechos reservados
